Preliminaries
Consider the common analysis of a neuroimaging experiment. At each voxel, vertex, face or edge (or any other imaging unit), we have a linear model expressed as:
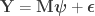
where 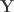 contains the experimental data,
contains the experimental data, 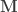 contains the regressors,
contains the regressors, 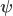 the regression coefficients, which are to be estimated, and
the regression coefficients, which are to be estimated, and  the residuals. For a linear null hypothesis
the residuals. For a linear null hypothesis  , where
, where 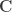 is a contrast. If
is a contrast. If  , the Student’s t statistic can be calculated as:
, the Student’s t statistic can be calculated as:
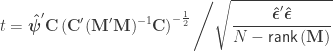
where the hat on 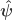 and
and 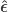 indicate that these are quantities estimated from the sample. If
indicate that these are quantities estimated from the sample. If 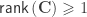 , the F statistic can be obtained as:
, the F statistic can be obtained as:
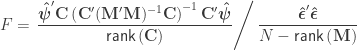
When , 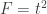 . For either of these statistics, we can assess their significance by repeating the same fit after permuting or (i.e., a permutation test), or by referring to the formula for the distribution of the corresponding statistic, which is available in most statistical software packages (i.e., a parametric test).
. For either of these statistics, we can assess their significance by repeating the same fit after permuting or (i.e., a permutation test), or by referring to the formula for the distribution of the corresponding statistic, which is available in most statistical software packages (i.e., a parametric test).
Permutation tests don’t depend on the same assumptions on which parametric tests are based. As some of these assumptions can be quite stringent in practice, permutation methods arguably should be preferred as a general framework for the statistical analysis of imaging data. At each permutation, a new statistic is computed and used to build its empirical distribution from which the p-values are obtained. In practice it’s not necessary to build the full distribution, and it suffices to increment a counter at each permutation. At the end, the counter is divided by the number of permutations to produce a p-value.
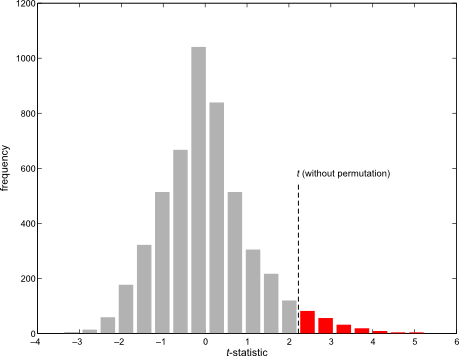
An example of a permutation distribution
Using permutation tests, correction for multiple testing using the family-wise error rate (fwer) is trivial: rather than build the permutation distribution at each voxel, a single distribution of the global maximum of the statistic across the image is constructed. Each permutation yields one maximum, that is used to build the distribution. Any dependence between the tests is implicitly captured, with not need to model it explicitly, nor to introduce even more assumptions, a problem that hinders methods such as the random field theory.
Exchangeability blocks
Permutation is allowed if it doesn’t affect the joint distribution of the error terms, i.e., if the errors are exchangeable. Some types of experiments may involve repeated measurements or other kinds of dependency, such that exchangeability cannot be guaranteed between all observations. However, various cases of structured dependency can still be accommodated if sets (blocks) of observations are shuffled as a whole, or if shuffling happens only within set (block). It is not necessary to know or to model the exact dependence structure between observations, which is captured implicitly as long as the blocks are defined correctly.
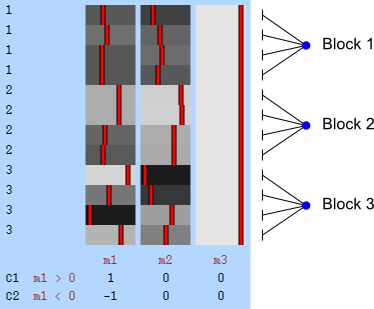
Permutation within block.
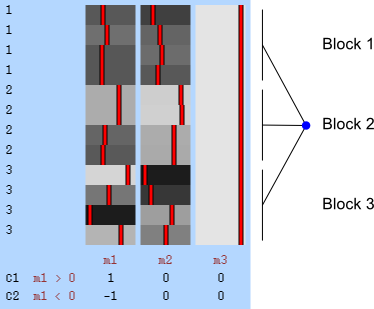
Permutation of blocks as a whole.
The two figures above are of designs constructed using the fsl software package. In fsl, within-block permutation is available in randomise with the option -e, used to supply a file with the definition of blocks. For whole-block permutation, in addition to the option -e, the option --permuteBlocks needs to be supplied.
The G-statistic
The presence of exchangeability blocks solves a problem, but creates another. Having blocks implies that observations may not be pooled together to produce a non-linear parameter estimate such as the variance. In other words: the mere presence of exchangeability blocks, either for shuffling within or as a whole, implies that the variances may not be the same across all observations, and a single estimate of this variance is likely to be inaccurate whenever the variances truly differ, or if the groups don’t have the same size. This also means that the F or t statistics may not behave as expected.
The solution is to use the block definitions and the permutation strategy is to define groups of observations that are known or assumed to have identical variances, and pool only the observations within group for variance estimation, i.e., to define variance groups (vgs).
The F-statistic, however, doesn’t allow such multiple groups of variances, and we need to resort to another statistic. In Winkler et al. (2014) we propose:
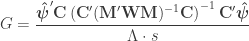
where  is a diagonal matrix that has elements:
is a diagonal matrix that has elements:
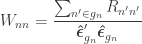
and where  are the
are the  diagonal elements of the residual forming matrix, and
diagonal elements of the residual forming matrix, and 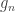 is the variance group to which the
is the variance group to which the 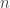 -th observation belongs. The remaining denominator term,
-th observation belongs. The remaining denominator term,  , is given by (Welch, 1951):
, is given by (Welch, 1951):
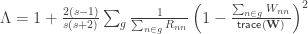
where 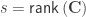 . The matrix can be seen as a weighting matrix, the square root of which normalises the model such that the errors have then unit variance and can be ignored. It can also be seen as being itself a variance estimator. In fact, it is the very same variance estimator proposed by Horn et al (1975).
. The matrix can be seen as a weighting matrix, the square root of which normalises the model such that the errors have then unit variance and can be ignored. It can also be seen as being itself a variance estimator. In fact, it is the very same variance estimator proposed by Horn et al (1975).
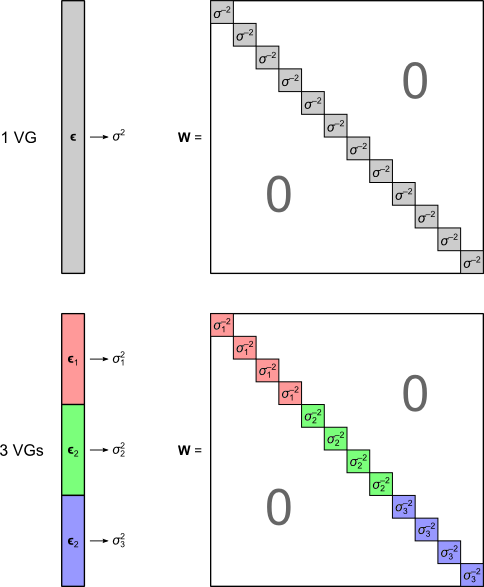
The W matrix used with the G statistic. It is constructed from the estimated variances of the error terms.
The matrix has a crucial role in making the statistic pivotal in the presence of heteroscedasticity. Pivotality means that the statistic has a sampling distribution that is not dependent on any unknown parameter. For imaging experiments, it’s important that the statistic has this property, otherwise correction for multiple testing that controls fwer will be inaccurate, or possibly invalid altogether.
When  , the t-equivalent to the G-statistic is
, the t-equivalent to the G-statistic is 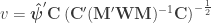 , which is the well known Aspin-Welch
, which is the well known Aspin-Welch  -statistic for the Behrens-Fisher problem. The relationship between and G is the same as between t and F, i.e., when the rank of the contrast equals to one, the latter is simply the square of the former. The G statistic is a generalization of all these, and more, as we show in the paper, and summarise in the table below:
-statistic for the Behrens-Fisher problem. The relationship between and G is the same as between t and F, i.e., when the rank of the contrast equals to one, the latter is simply the square of the former. The G statistic is a generalization of all these, and more, as we show in the paper, and summarise in the table below:
|
|
 |
| Homoscedastic errors, unrestricted exchangeability |
Square of Student’s t |
F-ratio |
| Homoscedastic within vg, restricted exchangeability |
Square of Aspin-Welch |
Welch’s  |
In the absence of variance groups (i.e., all observations belong to the same vg), G and are equivalent to F and t respectively.
Although not typically necessary if permutation methods are to be preferred, approximate parametric p-values for the G-statistic can be computed from an F-distribution with 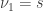 and
and  .
.
While the error rates are controlled adequately (a feature of permutation tests in general), the G-statistic offers excellent power when compared to the F-statistic, even when the assumptions of the latter are perfectly met. Moreover, by preserving pivotality, it is an adequate statistic to control of the error rate in the in the presence of multiple tests.
In this post, the focus is in using G for imaging data, but of course, it can be used for any dataset in which a linear model where variances cannot be assumed to be the same is used, i.e., when heteroscedasticity is or could be present.
Note that the G-statistic has nothing to do with the G-test. It is named as this for being a generalisation over various tests, including the commonly used t and F tests, as shown above.
Main reference
The core reference and results for the G-statistic have just been published in Neuroimage:
Other references
The two other references cited, which are useful to understand the variance estimator and the parametric approximation are:

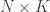
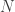
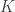


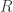
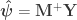


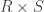


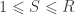


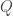



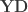
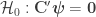
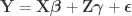


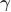

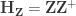


![\left[ \mathbf{X} \; \mathbf{Z} \right]](https://s0.wp.com/latex.php?latex=%5Cleft%5B+%5Cmathbf%7BX%7D+%5C%3B+%5Cmathbf%7BZ%7D+%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
![\boldsymbol{\psi} = \left[ \boldsymbol{\beta}' \; \boldsymbol{\gamma}' \right]'](https://s0.wp.com/latex.php?latex=%5Cboldsymbol%7B%5Cpsi%7D+%3D+%5Cleft%5B+%5Cboldsymbol%7B%5Cbeta%7D%27+%5C%3B+%5Cboldsymbol%7B%5Cgamma%7D%27+%5Cright%5D%27&bg=ffffff&fg=333333&s=0&c=20201002)
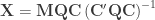



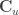

![[\mathbf{C} \; \mathbf{C}_u]](https://s0.wp.com/latex.php?latex=%5B%5Cmathbf%7BC%7D+%5C%3B+%5Cmathbf%7BC%7D_u%5D&bg=ffffff&fg=333333&s=0&c=20201002)
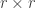
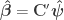

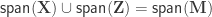



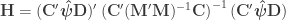
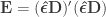

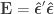
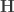
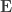

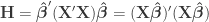
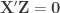

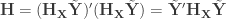

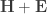


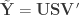

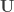






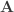

 distinct possible orderings of these cups, and by telling the subject in advance that there are four cups of each type, this guarantees that the answer will include four of each.
distinct possible orderings of these cups, and by telling the subject in advance that there are four cups of each type, this guarantees that the answer will include four of each.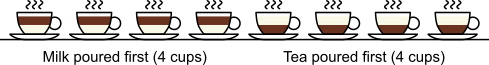
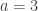


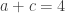
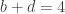
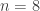
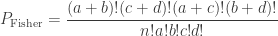
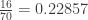 . This is not the final result, though: what matters to disprove the hypothesis that she is not able to discriminate is how likely it would be for her to find a result at least as extreme as the one observed. In this case, there is one case that is more extreme, which would be the one in which she would have made correct guesses for all the 8 cups, in which case the values in the contingency table above would have been
. This is not the final result, though: what matters to disprove the hypothesis that she is not able to discriminate is how likely it would be for her to find a result at least as extreme as the one observed. In this case, there is one case that is more extreme, which would be the one in which she would have made correct guesses for all the 8 cups, in which case the values in the contingency table above would have been 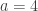 ,
,  ,
,  , and
, and  , with a probability computed with the same formula as
, with a probability computed with the same formula as  . Adding these two probabilities together yield
. Adding these two probabilities together yield  .
.
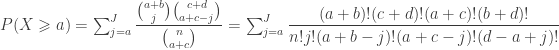
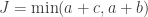 . Computing from the above (details omitted), yield the same value as using Fisher’s presentation, that is, the p-value is (exactly) 0.24286.
. Computing from the above (details omitted), yield the same value as using Fisher’s presentation, that is, the p-value is (exactly) 0.24286.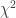 method
method
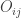 is the observed value for the element in the position
is the observed value for the element in the position 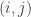 in the table,
in the table, 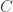 are respectively the number of rows and columns, and
are respectively the number of rows and columns, and 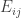 is the expected value for these elements if the null hypothesis is true. The values
is the expected value for these elements if the null hypothesis is true. The values 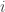 and column
and column 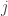 , divided by the overall number of observations
, divided by the overall number of observations 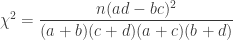
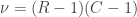 .
. , which corresponds to a p-value of 0.07865. However, it is well known that this method is inaccurate if cells in the table have too small quantities, usually below 5 or 6.
, which corresponds to a p-value of 0.07865. However, it is well known that this method is inaccurate if cells in the table have too small quantities, usually below 5 or 6.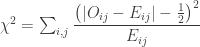
 , and a p-value of 0.23975, which is very similar to the one given by the Fisher method. Note again that this approach, like the
, and a p-value of 0.23975, which is very similar to the one given by the Fisher method. Note again that this approach, like the  be a column vector containing binary indicators for whether milk was truly poured first. Let
be a column vector containing binary indicators for whether milk was truly poured first. Let  be a column vector containing binary indicators for whether the lady answered that milk was poured first. The
be a column vector containing binary indicators for whether the lady answered that milk was poured first. The  , where
, where  is a regression coefficient, and
is a regression coefficient, and 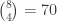 possible unique rearrangements. Out of these, in 17, there are 6 or more (out of 8) correct answers matching the values in
possible unique rearrangements. Out of these, in 17, there are 6 or more (out of 8) correct answers matching the values in 

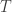 traits,
traits, 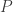 covariates and
covariates and 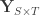 are the observed trait values for each subject,
are the observed trait values for each subject,  is a matrix of covariates,
is a matrix of covariates, 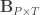 is a matrix of unknown covariates’ weights, and
is a matrix of unknown covariates’ weights, and  are the residuals after the covariates have been taken into account.
are the residuals after the covariates have been taken into account.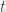 of
of  are assumed to follow a multivariate normal distribution
are assumed to follow a multivariate normal distribution  , where
, where  is the between-subject covariance matrix. The elements of each row
is the between-subject covariance matrix. The elements of each row  of
of 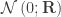 , where
, where  is the between-trait covariance matrix. Both
is the between-trait covariance matrix. Both 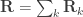 and
and 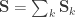 . For a discussion on these equalities, see Eisenhart (1947) [see references at the end].
. For a discussion on these equalities, see Eisenhart (1947) [see references at the end]. be the stacked vector of traits,
be the stacked vector of traits,  is the matrix of covariates,
is the matrix of covariates,  the vector with the covariates’ weights,
the vector with the covariates’ weights,  the residuals after the covariates have been taken into account, and
the residuals after the covariates have been taken into account, and 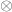 represent the
represent the 
 is assumed to follow a multivariate normal distribution
is assumed to follow a multivariate normal distribution  , where
, where  can be seen as the sum of
can be seen as the sum of 
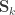 can be modelled as correlation matrices. The associated scalars are absorbed into the (to be estimated)
can be modelled as correlation matrices. The associated scalars are absorbed into the (to be estimated)  .
. ![\mathbf{R} = \left[ \begin{array}{ccc} \mathsf{Var}(\upsilon_1) & \cdots & \mathsf{Cov}(\upsilon_1,\upsilon_T) \\ \vdots & \ddots & \vdots \\ \mathsf{Cov}(\upsilon_T,\upsilon_1) & \cdots & \mathsf{Var}(\upsilon_T) \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7BR%7D+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BCov%7D%28%5Cupsilon_1%2C%5Cupsilon_T%29+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cmathsf%7BCov%7D%28%5Cupsilon_T%2C%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
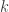 -th component:
-th component:![\mathbf{R}_k = \left[ \begin{array}{ccccc} \mathsf{Var}_k(\upsilon_1) & \cdots & \mathsf{Cov}_k(\upsilon_1,\upsilon_T) \\ \vdots & \ddots & \vdots \\ \mathsf{Cov}_k(\upsilon_T,\upsilon_1) & \cdots & \mathsf{Var}_k(\upsilon_T) \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7BR%7D_k+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccccc%7D+%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BCov%7D_k%28%5Cupsilon_1%2C%5Cupsilon_T%29+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cmathsf%7BCov%7D_k%28%5Cupsilon_T%2C%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
 as:
as:![\mathbf{\mathring{R}} = \left[ \begin{array}{ccc} \frac{\mathsf{Var}(\upsilon_1)}{\mathsf{Var}(\upsilon_1)} & \cdots & \frac{\mathsf{Cov}(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}(\upsilon_1)\mathsf{Var}(\upsilon_T)\right)^{1/2}} \\ \vdots & \ddots & \vdots \\ \frac{\mathsf{Cov}(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}(\upsilon_1)\mathsf{Var}(\upsilon_T)\right)^{1/2}} & \cdots & \frac{\mathsf{Var}(\upsilon_T)}{\mathsf{Var}(\upsilon_T)} \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5Cmathring%7BR%7D%7D+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cfrac%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BCov%7D%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cfrac%7B%5Cmathsf%7BCov%7D%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%7D+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
![\mathbf{\mathring{R}}_k = \left[ \begin{array}{ccc} \frac{\mathsf{Var}_k(\upsilon_1)}{\mathsf{Var}(\upsilon_1)} & \cdots & \frac{\mathsf{Cov}_k(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}(\upsilon_1)\mathsf{Var}(\upsilon_T)\right)^{1/2}} \\ \vdots & \ddots & \vdots \\ \frac{\mathsf{Cov}_k(\upsilon_T,\upsilon_1)}{\left(\mathsf{Var}(\upsilon_T)\mathsf{Var}(\upsilon_1)\right)^{1/2}} & \cdots & \frac{\mathsf{Var}_k(\upsilon_T)}{\mathsf{Var}(\upsilon_T)} \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5Cmathring%7BR%7D%7D_k+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_T%2C%5Cupsilon_1%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cright%29%5E%7B1%2F2%7D%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%7D+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
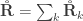 holds. The diagonal elements of
holds. The diagonal elements of  may receive particular names, e.g., heritability, environmentability, dominance effects, shared enviromental effects, etc., depending on what is modelled in the corresponding
may receive particular names, e.g., heritability, environmentability, dominance effects, shared enviromental effects, etc., depending on what is modelled in the corresponding 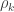 that correspond, e.g. to the genetic or environmental correlation. These off-diagonal elements are instead the signed
that correspond, e.g. to the genetic or environmental correlation. These off-diagonal elements are instead the signed  when
when 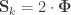 , or their
, or their  -equivalent for other variance components (see below). In this particular case, they can also be called “bivariate heritabilities” (Falconer and MacKay, 1996). A matrix
-equivalent for other variance components (see below). In this particular case, they can also be called “bivariate heritabilities” (Falconer and MacKay, 1996). A matrix  that contains these correlations
that contains these correlations ![\mathbf{\breve{R}}_k = \left[ \begin{array}{ccc} \frac{\mathsf{Var}_k(\upsilon_1)}{\mathsf{Var}_k(\upsilon_1)} & \cdots & \frac{\mathsf{Cov}_k(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}_k(\upsilon_1)\mathsf{Var}_k(\upsilon_T)\right)^{1/2}} \\ \vdots & \ddots & \vdots \\ \frac{\mathsf{Cov}_k(\upsilon_T,\upsilon_1)}{\left(\mathsf{Var}_k(\upsilon_T)\mathsf{Var}_k(\upsilon_1)\right)^{1/2}} & \cdots & \frac{\mathsf{Var}_k(\upsilon_T)}{\mathsf{Var}_k(\upsilon_T)} \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5Cbreve%7BR%7D%7D_k+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%7D%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_T%2C%5Cupsilon_1%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%5Cright%29%5E%7B1%2F2%7D%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%7D%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%7D+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
 , the coefficient of familial relationship between subjects, and
, the coefficient of familial relationship between subjects, and 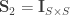 . In this case, the
. In this case, the  represent the heritability (
represent the heritability ( ) for each trait
) for each trait  contains
contains  , the environmentability. The off-diagonal elements of
, the environmentability. The off-diagonal elements of  are the off-diagonal elements of
are the off-diagonal elements of 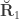 , whereas the off-diagonal elements of
, whereas the off-diagonal elements of 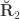 are
are 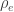 , the environmental correlations between traits. In this particular case, the components of
, the environmental correlations between traits. In this particular case, the components of 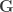 and
and 
 and rearranging the terms gives:
and rearranging the terms gives: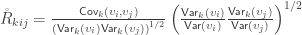
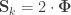 , the above reduces to
, the above reduces to  , which is the signed version of
, which is the signed version of  when
when 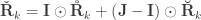
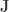 is a matrix of ones,
is a matrix of ones,  is the identity, both of size
is the identity, both of size 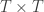 , and
, and 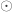 is the
is the 


 is the number of observations on the stacked vector
is the number of observations on the stacked vector 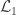 ), the loglikelihood of a model in which the parameters being tested are constrained to zero, the null model (
), the loglikelihood of a model in which the parameters being tested are constrained to zero, the null model (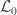 ). The statistic is given by
). The statistic is given by  (Wilks, 1938), which here is asymptotically distributed as a 50:50 mixture of a
(Wilks, 1938), which here is asymptotically distributed as a 50:50 mixture of a  and
and 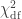 distributions, where df is the number of parameters being tested and free to vary in the unconstrained model (Self and Liang, 1987). From this distribution the p-values can be obtained.
distributions, where df is the number of parameters being tested and free to vary in the unconstrained model (Self and Liang, 1987). From this distribution the p-values can be obtained. , is probably the most interesting. It gives a probabilistic estimate that a random gene from a given subject
, is probably the most interesting. It gives a probabilistic estimate that a random gene from a given subject 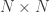 matrix termed kinship matrix, usually represented as
matrix termed kinship matrix, usually represented as  , that has elements
, that has elements 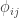 , and that can be used to model the covariance between individuals in quantitative genetics.
, and that can be used to model the covariance between individuals in quantitative genetics.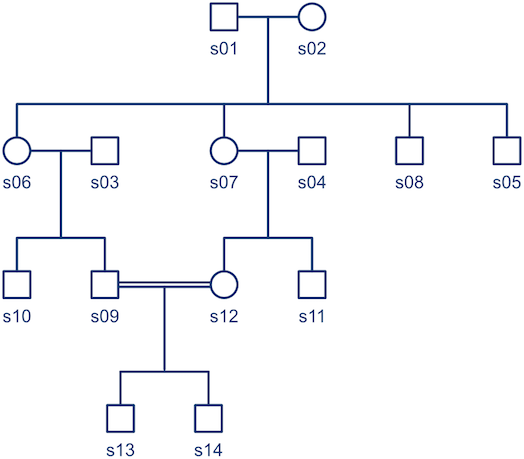
 ), is:
), is: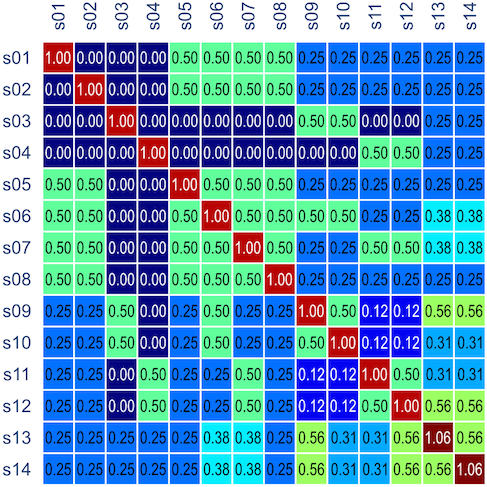

 . Each individual has two copies, one from paternal, another from maternal origin; these can be indicated as
. Each individual has two copies, one from paternal, another from maternal origin; these can be indicated as  and
and  for individual
for individual 
 , a respective probability
, a respective probability  can be assigned; these are called coefficients of identity by descent. These probabilities can be calculated at every generation following very elementary rules. For most problems, however, the distinction between paternal and maternal origin of a gene is irrelevant, and some of the above states are equivalent to others. If these are condensed, we can retain 9 distinct ways, shown in the figure below:
can be assigned; these are called coefficients of identity by descent. These probabilities can be calculated at every generation following very elementary rules. For most problems, however, the distinction between paternal and maternal origin of a gene is irrelevant, and some of the above states are equivalent to others. If these are condensed, we can retain 9 distinct ways, shown in the figure below:
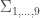 , a respective probability
, a respective probability 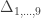 can be assigned; these are called condensed coefficients of identity by descent, and relate to the former as:
can be assigned; these are called condensed coefficients of identity by descent, and relate to the former as: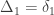
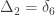

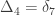




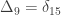
 ,
,  and
and 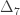 correspond to his coefficients
correspond to his coefficients  ,
,  and
and  .
.
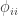 is the kinship of a subject with himself. Two genes taken from the same individual can either be the same gene (probability
is the kinship of a subject with himself. Two genes taken from the same individual can either be the same gene (probability  of being the same) or be the genes inherited from father and mother, in which case the probability is given by the coefficient of kinship between the parents. In other words,
of being the same) or be the genes inherited from father and mother, in which case the probability is given by the coefficient of kinship between the parents. In other words,  . If both parents are unrelated,
. If both parents are unrelated, 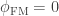 , such that the kinship of a subject with himself is
, such that the kinship of a subject with himself is 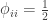 .
. (see below about the coefficient of inbreeding,
(see below about the coefficient of inbreeding,  ). Thus, if there are
). Thus, if there are  generations between
generations between  . If
. If 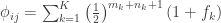
 , and used to model the covariance between subjects as
, and used to model the covariance between subjects as  (
(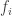 can be computed from the coefficients of identity:
can be computed from the coefficients of identity:

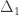

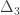
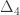

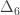
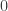
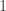

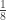
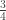

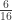






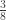

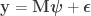
 vector of observations,
vector of observations,  matrix of explanatory variables,
matrix of explanatory variables,  vector of regression coefficients, and
vector of regression coefficients, and  , where
, where  matrix that defines a contrast of regression coefficients, satisfying
matrix that defines a contrast of regression coefficients, satisfying 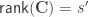 and
and  .
.
 vector of observations,
vector of observations, 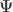 is the
is the 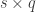 vector of regression coefficients, and
vector of regression coefficients, and  , where
, where  matrix that defines a contrast of observed variables, satisfying
matrix that defines a contrast of observed variables, satisfying  and
and 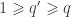 .
.
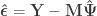
 and
and 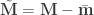 .
.
 statistic as:
statistic as: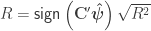
 statistic can be computed as:
statistic can be computed as: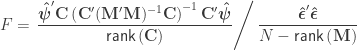
 , and
, and 

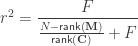
 is known, the formula can be solved for
is known, the formula can be solved for 

![\mathbf{A} = \left[\mathbf{y}\; \mathbf{M}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7BA%7D+%3D+%5Cleft%5B%5Cmathbf%7By%7D%5C%3B+%5Cmathbf%7BM%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002) , and
, and  be the inverse of the correlation matrix of the columns of
be the inverse of the correlation matrix of the columns of 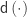 the diagonal operator, that returns a column vector with the diagonal entries of a square matrix. Then the matrix with the partial correlations is:
the diagonal operator, that returns a column vector with the diagonal entries of a square matrix. Then the matrix with the partial correlations is:
 ” is taken elementwise (i.e., not matrix power).
” is taken elementwise (i.e., not matrix power).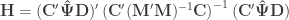 as the sums of products of the hypothesis. In fact, the original model can be modified as
as the sums of products of the hypothesis. In fact, the original model can be modified as  , where
, where 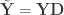 ,
, 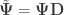 and
and  .
. , this is an univariate model, otherwise it remains multivariate, although
, this is an univariate model, otherwise it remains multivariate, although  .
.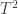 statistic can be computed as:
statistic can be computed as:


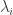 be the eigenvalues of
be the eigenvalues of 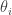 the eigenvalues of
the eigenvalues of  . Then:
. Then: .
. .
. .
.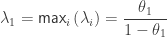 (analogous to
(analogous to  (analogous to
(analogous to  is the
is the 






 is the smoothed quantity at the vertex or face
is the smoothed quantity at the vertex or face  is the quantity assigned to the
is the quantity assigned to the  vertices or faces,
vertices or faces, 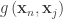 is the geodesic distance between vertices or faces with coordinates
is the geodesic distance between vertices or faces with coordinates 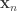 and
and  , and
, and  is the Gaussian filter, defined as a function of the geodesic distance between points.
is the Gaussian filter, defined as a function of the geodesic distance between points. are known and organised into a distance matrix
are known and organised into a distance matrix  , with the values at each row scaled so as to add up to unity, and the same smoothing can proceed as a simple matrix multiplication:
, with the values at each row scaled so as to add up to unity, and the same smoothing can proceed as a simple matrix multiplication:
 from the filter center, in a sphere of radius
from the filter center, in a sphere of radius 
 . Even using sparse matrices, this may require a large amount of memory space. For practical purposes, a filter with width
. Even using sparse matrices, this may require a large amount of memory space. For practical purposes, a filter with width