How about processing brain imaging data on a Raspberry Pi? The different versions of this little device have performed exceptionally well for education, entertainment, and for a variety of do-it-yourself projects, with many examples listed in websites such as Instructables and Adafruit. Most of these applications are not computationally as intensive. Yet, the small size, low power consumption, improved hardware in recent models, and low price, may make this feasible.
The Pi 2
 Released earlier this year, the Raspberry Pi 2 (Model B) features a quad-core 900 MHz ARM processor, 1 GB of RAM, GPU, 4 USB ports, 10/100 Mbps Ethernet, HDMI and audio outputs, camera and display ports, as well as a low level general purpose interface (GPIO), all in a portable board of 85.6 mm × 56.5 mm (the same size as a credit card). It is powered by a 5 V, 800 mA DC (4 W) source, and sold for £30 or less.
Released earlier this year, the Raspberry Pi 2 (Model B) features a quad-core 900 MHz ARM processor, 1 GB of RAM, GPU, 4 USB ports, 10/100 Mbps Ethernet, HDMI and audio outputs, camera and display ports, as well as a low level general purpose interface (GPIO), all in a portable board of 85.6 mm × 56.5 mm (the same size as a credit card). It is powered by a 5 V, 800 mA DC (4 W) source, and sold for £30 or less.
Differently than earlier models, which had a CPU based on the ARMv6, the Pi 2 uses an ARM Cortex-A7 processor, which on its turn based on the ARMv7 architecture. Although there are Linux distributions that can run on the earlier models (such as ports of Debian, openSUSE, and Fedora), this change widens potential applications, not only because there are more ports available for ARMv7 (e.g., openSUSE, Debian, CentOS, among others), but also, the higher performance suggests that somewhat heavier data processing can be considered.
It is also possible to assemble multiple Pis in a cluster, using distributed computing engines such as SLURM, TORQUE or SGE. The Pi has the core requisites: it runs on Linux and comes with a decent 10/100 Mbps Ethernet port, such that creating a system is a matter of assembling the pieces and configuring.
Neuroimaging with a small footprint
With this relatively high amount of computing power in such a physically small size and affordable price, the question is immediate: It is feasible to do neuroimaging on the Pi? The availability of Linux distributions for ARM platforms suggest that yes. However, the binaries for imaging software distributed for popular platforms as x86 (i386) and x86-64 (amd64) cannot work directly. Rather, the applications would need to be compiled from source.
 For the FMRIB Software Library (FSL), the source code can be downloaded and the compilation proceed. Much simpler than that, however, is to take a different route: FSL has been included in NeuroDebian. This alone does not seem helpful, as the packages in the repository are only for 32-bit and 64-bit PCs (the i386 and amd64 ports), and SPARC. However, these packages have made into the upstream Debian, which means they are available for all platforms for which Debian itself has been ported. This includes the ARMv7 that powers the Pi, for which the port armhf (for chips that use a hardware floating point unit) can be used.
For the FMRIB Software Library (FSL), the source code can be downloaded and the compilation proceed. Much simpler than that, however, is to take a different route: FSL has been included in NeuroDebian. This alone does not seem helpful, as the packages in the repository are only for 32-bit and 64-bit PCs (the i386 and amd64 ports), and SPARC. However, these packages have made into the upstream Debian, which means they are available for all platforms for which Debian itself has been ported. This includes the ARMv7 that powers the Pi, for which the port armhf (for chips that use a hardware floating point unit) can be used.
The steps to have a working installation of FSL on the Pi 2 are described below. Other interesting software, such as FreeSurfer, would need to be compiled from the source. For SPM, there is no Matlab port at the moment, but Octave runs without problems, such that most functionalities are expected to work. Applications based on Java, such as Mango, work without problems.
Requirements
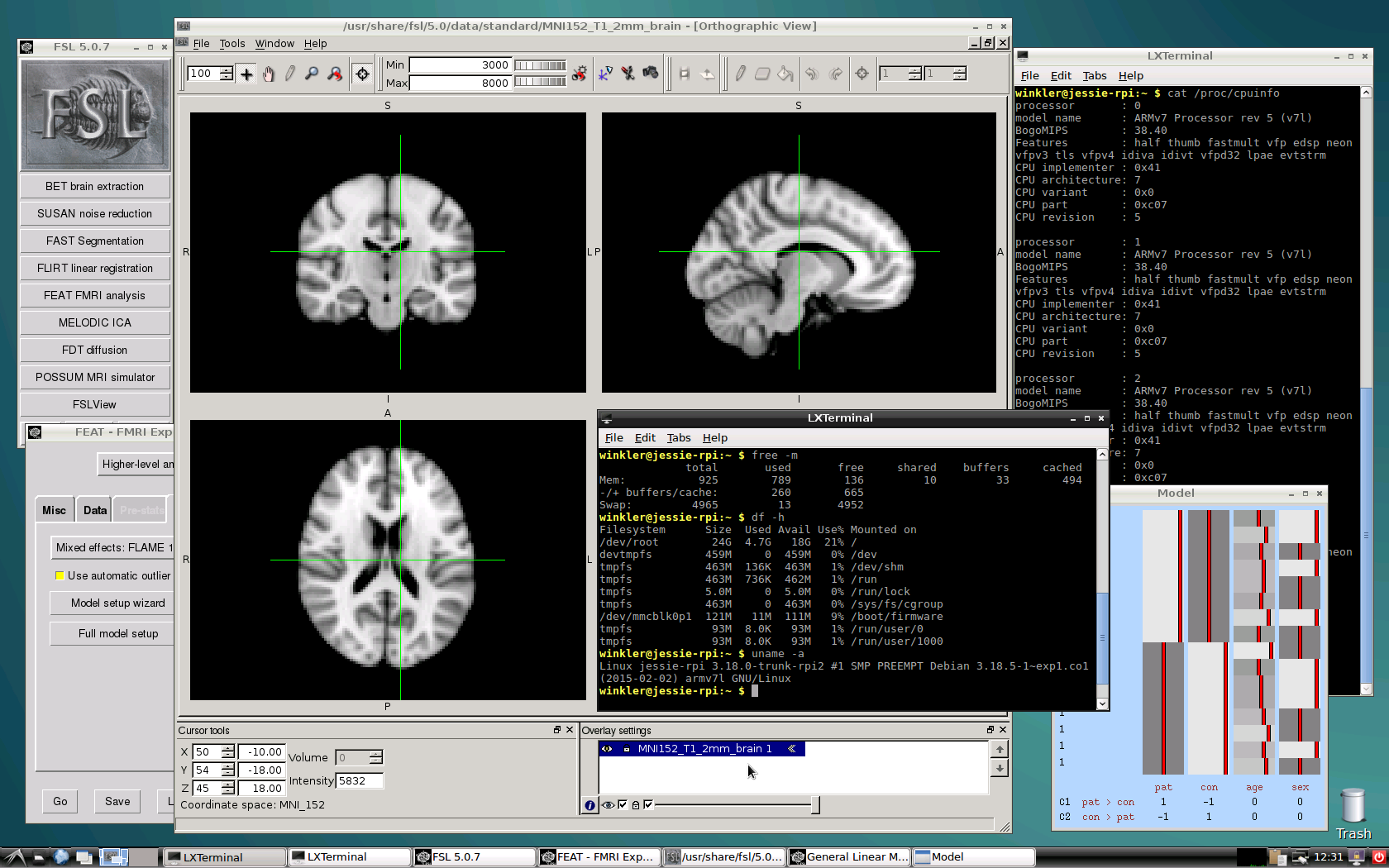
The photo at the top shows the hardware assembly used for this article. The following is required:
- A Raspberry Pi 2 (Model B).
- Power source (can be the USB port of another computer).
- Micro SD card with at least 8 GB (below it is assumed 32 GB).
- Ethernet cable and a network that provides internet access.
- Optional: HDMI display and cable, USB keyboard, and possibly a USB mouse if a graphical system will be installed. Alternatively, a headless system also works, with access via SSH. Below it is assumed a display is connected.
The procedure
Step 1: Download the system image kindly prepared by Sjoerd Simons, and uncompress it:
wget https://images.collabora.co.uk/rpi2/jessie-rpi2-20150705.img.gz
gunzip jessie-rpi2-20150705.img.gz
This image contains only a minimal set of Debian Jessie packages. It uses the kernel 3.18.5, and received a few firmware and boot tweaks that are specific to the Pi 2.
Step 2: Use your favourite utility to transfer the image to the micro SD card. For example, using Linux, run the following, replacing /dev/sdX for the letter corresponding to your SD card (warning: this will erase all data stored in the card):
dd bs=1M if=jessie-rpi2-20150705.img of=/dev/sdX
In some systems, the card may be in /dev/mmcblk0 instead in /dev/sdX. If a Linux machine is not available, but instead a Mac or even Windows, the instructions to install Raspbian also apply.
Step 3: Insert the card in the Pi and boot the system. In this image, the default root password is debian (you can change it for something sensible).
Step 4: The main partition in this disk image (mounted as /) has only 2.6 GB, which is not enough. Also, often more than 1 GB of memory is needed, so swap space for virtual memory is necessary. Use fdisk (as root) to expand the main partition and to create a new partition for swap. Usually this is done interactively. If the card has exactly 32 GB, the line below can be used directly, bypassing the interactive mode. It will define a main partition with 24 GB, and the remaining, about 5 GB, will be left for swap. For cards of different sizes, run fdisk manually, or change the line below accordingly.
printf "d\n2\nn\np\n\n\n+24G\nn\np\n\n\n\nt\n3\n82\nw\n" | fdisk -uc /dev/mmcblk0
Note that, when seen from the Pi, the SD card is at /dev/mmcblk0, not /dev/sdX.
Reboot (shutdown -r now), then after logging in again, run:
resize2fs /dev/mmcblk0p2
mkswap /dev/mmcblk0p3
swapon /dev/mmcblk0p3
The swapon command enables the swap partition for immediate use. To make the change permanent for the next reboot, edit the file /etc/fstab adding:
/dev/mmcblk0p3 swap swap defaults 0 0
Step 5: Edit the /etc/apt/sources.list so as to include the official Debian packages (you can replace the server for your favourite/closer mirror):
deb http://ftp.uk.debian.org/debian/ jessie main contrib non-free
Step 6: Refresh the cached list of packages, then install FSL:
apt-get update
apt-get install fsl
Step 7: The installation is almost ready. The downloaded packages do not have the “data” directory of FSL, which contains the atlases and standard space images. To obtain these, do one of the following:
- From a separate FSL installation (e.g., from a different computer), copy the contents of the
${FSLDIR}/data to the /usr/share/fsl/data of the newly installed system on the Raspberry Pi. This can be done over the network, via ssh, or after plugging in and mounting (with the correct privileges) the card in a different Linux system.
- If another computer with FSL installed is not available, download FSL for CentOS or Mac (at the end of the downloads page, under “Advanced Users”), then uncompress the downloaded file, and copy the whole contents of the
data directory to /usr/share/fsl/data of the Pi via ssh
- If another computer is not at all accessible for this step, these files can be obtained using the Pi itself, from the command line. Logged in as root in the newly installed system, run:
cd /usr/share
wget -O- http://fsl.fmrib.ox.ac.uk/fsldownloads/fsl-5.0.9-centos6_64.tar.gz | tar xzfv - fsl/data
- A last option is to skip this step, go to Step 9 below, then download and copy using a graphical web-browser from an installed desktop environment.
Step 8: Add this line to the file ~/.profile:
. /etc/fsl/fsl.sh
That’s it. All that is needed to run FSL from the command line has been done.
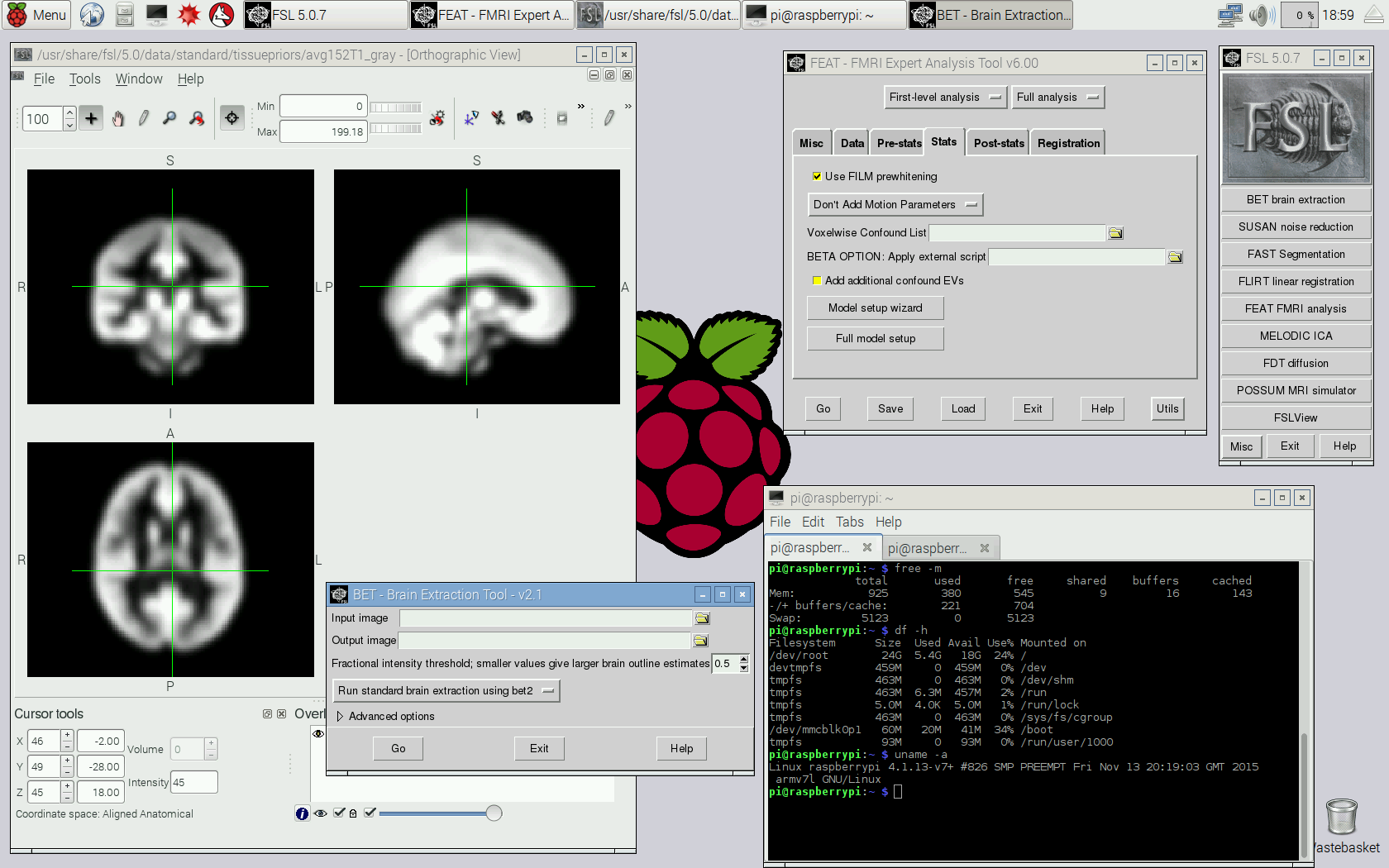
Step 9 (optional): The installation up to this step does not include a graphical user interface. To have one, install X and a desktop environment. For lightweight options, LXDE or XFCE can be considered. A screenshot of LXDE with FSLview and two terminal windows showing some system information is below (usually one would not run as root, but create an user account).

Using Raspbian
The official operating system for the Raspberry Pi, Raspbian, is a customised version of Debian, thus capable of running FSL directly. However, FSL is not in the official rpi repository. It can still be installed following similar steps as above, remembering to use sudo with commands require root privileges (the default account is rpi and the password is raspberry), and with care in the repartitioning, as the official disk image uses a different scheme. In Step 5, include the same Debian package source in the /etc/apt/sources.list file.

Overclocking
The Pi can be overclocked. Conservative, stable settings, that do not void the warranty, consist of increasing the CPU frequency to 1000 MHz (from the default 900), the GPU and SDRAM frequencies to 500 MHz (the defaults are 250 and 450 respectively), and the CPU/GPU voltage by 2 steps, i.e., by 2 × 25 mV, from 1.20 to 1.25 V. The overclock settings are adjusted in the file /boot/firmware/config.txt if using Debian (following the steps above), or in /boot/config.txt if using Raspbian:
arm_freq=1000
gpu_freq=500
sdram_freq=500
over_voltage=2
These settings cause the bogomips to jump from 38.40 to 64.00. The temperature of the onboard chips can increase, however, and a suggestion is to use heatsinks or fans, which are inexpensive and can be purchased online (fans would be powered by GPIO pins).
Performance
With the system up and running, it is time for some benchmarks. Although the assembly is exciting and in general the system speed respectable, unfortunately, processing using FEEDS suggests a poor performance. The table below compares the timings of the Pi 2 with default versus overclocked settings, relative to a minimal install of the Debian Jessie on a notebook with an Intel Core i5 processor and 8 GB of RAM.
|
Default settings |
Overclocked settings |
| PRELUDE & FUGUE |
6.0 |
4.8 |
| SUSAN |
15.1 |
11.9 |
| SIENAX |
13.3 |
10.4 |
| BET2 |
12.3 |
9.4 |
| FEAT |
12.1 |
9.6 |
| MELODIC |
15.9 |
12.2 |
| FIRST |
14.0 |
11.1 |
| FDT |
7.4 |
5.9 |
| FNIRT |
26.6 |
19.3 |
| Total time |
12.2 |
9.5 |
Running the whole FEEDS took 22 minutes in the Intel Core i5, whereas in the Pi 2 it took 4h29min with the default settings, and 3h30min after overclocking. It should be noted, however, that the 1 GB of RAM is not sufficient to run the test without using virtual memory (swapping). This needs to be taken into account when evaluating the table above. The SD card used for the tests is a Class 10, which is not as fast as actual RAM (faster cards would have their performance curtailed by hardware limits).
The performances of Debian and Raspbian on the Pi 2 are nearly identical. Running in the graphical mode (at least with LXDE) or in a console-only system do not seem to impact results, at least as far only one instance of FEEDS was running.
Conclusion
It is possible to run FSL on the Raspberry Pi 2, and the procedure is not too different than doing the same in an ordinary computer. The performance, however, suggests that the current model, being about ten times slower, may not be a competitive choice for brain imaging.

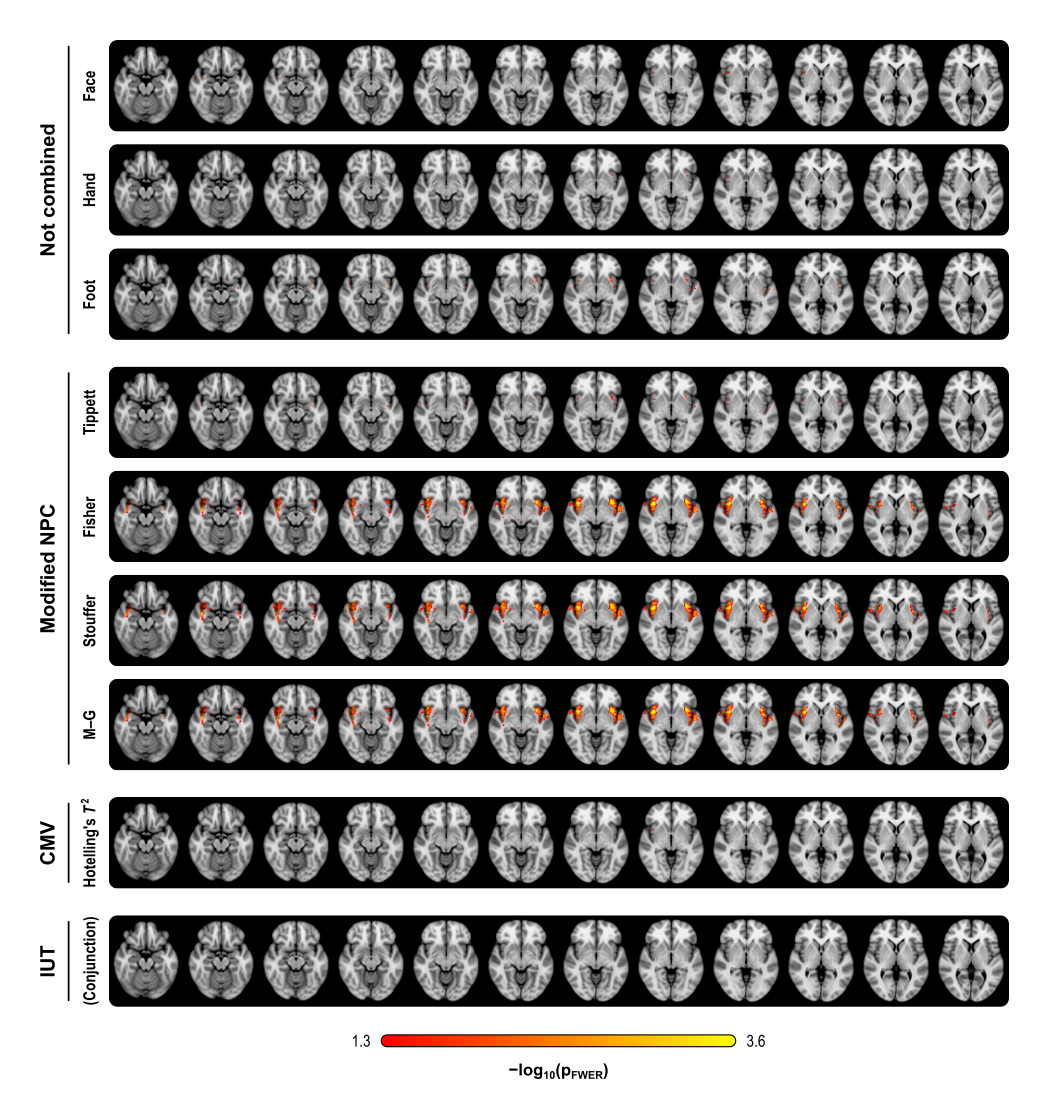
 , we expect, on average, to find 50 significant tests, even in the absence of any effect. This is known as the multiple testing problem. A review of the topic for brain imaging provided in Nichols and Hayasaka (2003) [see references at the end].
, we expect, on average, to find 50 significant tests, even in the absence of any effect. This is known as the multiple testing problem. A review of the topic for brain imaging provided in Nichols and Hayasaka (2003) [see references at the end]. ), or the p-values can be adjusted, such that instead of controlling the error rate at each individual test, the error rate is controlled for the whole set (family) of tests. Controlling such family-wise error rate (FWER) ensures that the chance of finding a significant result anywhere in the image is expected to be within a certain predefined level. For example, if there are 1000 voxels, and the FWER-adjusted test level is 0.05, we expect that, if the experiment is repeated for all the voxels 20 times, then on average in one of these repetitions there will be an error somewhere in the image. The adjustment of the p-values or of the test level is done using the distribution of the maximum statistic, something that most readers of this blog are certainly well aware of, as that permeates most of the imaging literature since the early 1990s.
), or the p-values can be adjusted, such that instead of controlling the error rate at each individual test, the error rate is controlled for the whole set (family) of tests. Controlling such family-wise error rate (FWER) ensures that the chance of finding a significant result anywhere in the image is expected to be within a certain predefined level. For example, if there are 1000 voxels, and the FWER-adjusted test level is 0.05, we expect that, if the experiment is repeated for all the voxels 20 times, then on average in one of these repetitions there will be an error somewhere in the image. The adjustment of the p-values or of the test level is done using the distribution of the maximum statistic, something that most readers of this blog are certainly well aware of, as that permeates most of the imaging literature since the early 1990s. voxels in an image. For a given voxel
voxels in an image. For a given voxel  ,
,  , with test statistic
, with test statistic 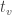 , the probability that
, the probability that 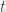 is denoted by:
is denoted by: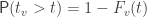
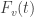 is the cumulative distribution function (cdf) of the test statistic. If the cutoff
is the cumulative distribution function (cdf) of the test statistic. If the cutoff  . With many voxels, the chances of this happening increase, even if there no effect is present. We can, however, adjust our cuttoff
. With many voxels, the chances of this happening increase, even if there no effect is present. We can, however, adjust our cuttoff  so that the probability of rejecting such family-wise null hypothesis remains within a certain level, say
so that the probability of rejecting such family-wise null hypothesis remains within a certain level, say  .
.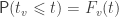 . The probability of the same happening for all voxels simultaneously is, therefore, simply the product of such probabilities, assuming of course that the voxels are all independent:
. The probability of the same happening for all voxels simultaneously is, therefore, simply the product of such probabilities, assuming of course that the voxels are all independent:
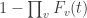 . If all voxels have identical distribution under the null, then the same is stated as
. If all voxels have identical distribution under the null, then the same is stated as  .
. . The random variable
. The random variable  is only smaller or equal than some cutoff
is only smaller or equal than some cutoff  .
. , the test in
, the test in  is considered significant if the joint tests using (1,2,3,4), (1,2,3), (1,2,4), (1,3,4), (1,2), (1,3), (1,4) and (1) are all significant (that is, all that include
is considered significant if the joint tests using (1,2,3,4), (1,2,3), (1,2,4), (1,3,4), (1,2), (1,3), (1,4) and (1) are all significant (that is, all that include 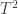 , MANOVA/MANCOVA, or NPC (Non-Parametric Combination), all of which are based on recomputing the test statistic from the original data, or others, based on the test statistics or p-values of each of the elementary
, MANOVA/MANCOVA, or NPC (Non-Parametric Combination), all of which are based on recomputing the test statistic from the original data, or others, based on the test statistics or p-values of each of the elementary  , which in imaging applications renders them unfeasible. However, there is one particular joint test that provides a direct algorithmic shortcut: using the
, which in imaging applications renders them unfeasible. However, there is one particular joint test that provides a direct algorithmic shortcut: using the  as the test statistic for the joint test. The maximum across all
as the test statistic for the joint test. The maximum across all 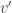 , with an even larger statistic (i.e.,
, with an even larger statistic (i.e., 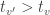 ), but the probability of such happening would not be captured by the distribution of a non-maximum. Hence the chance of finding a significant voxel anywhere in the image under the null hypothesis (the very definition of FWER) would not be controlled. Using the absolute maximum eliminates this logical leakage.
), but the probability of such happening would not be captured by the distribution of a non-maximum. Hence the chance of finding a significant voxel anywhere in the image under the null hypothesis (the very definition of FWER) would not be controlled. Using the absolute maximum eliminates this logical leakage.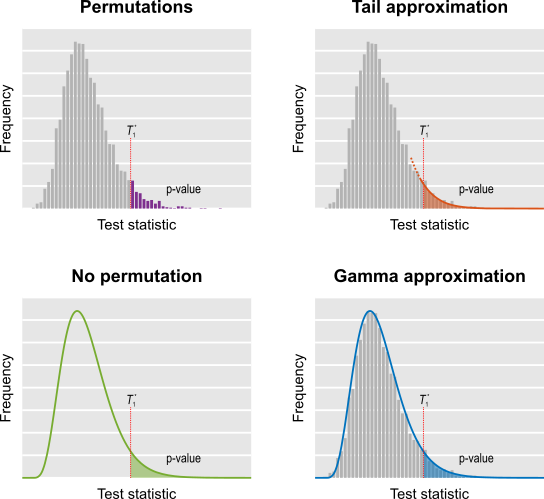





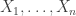 be a sequence of independent and identically distributed variables with cumulative distribution function (cdf)
be a sequence of independent and identically distributed variables with cumulative distribution function (cdf) 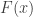 and let
and let 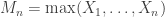 denote the maximum.
denote the maximum.
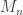 directly is problematic because as
directly is problematic because as  ,
, 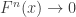 . Redefining the problem as a function of
. Redefining the problem as a function of 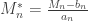 renders treatment simpler. The theorem can be stated then as: If there exist sequences of constants
renders treatment simpler. The theorem can be stated then as: If there exist sequences of constants  and
and  such that, as
such that, as 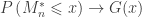
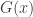 belongs to one of three “domains of attraction”:
belongs to one of three “domains of attraction”: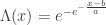 , for
, for 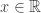 indicating that the distribution of
indicating that the distribution of  indicating that the distribution of
indicating that the distribution of 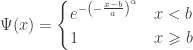 indicating that the distribution of
indicating that the distribution of 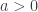 ,
, 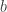 and, for families II and III,
and, for families II and III,  . To which of the three a particular
. To which of the three a particular  . Thus, we can infer about the asymptotic properties of the maximum while having only a limited knowledge of the properties of
. Thus, we can infer about the asymptotic properties of the maximum while having only a limited knowledge of the properties of  , whereas Fisher and Tippett used
, whereas Fisher and Tippett used 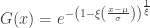
 , with parameters
, with parameters  (location),
(location),  (scale), and
(scale), and 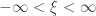 (shape). This is the generalised extreme value (GEV) family of distributions. If
(shape). This is the generalised extreme value (GEV) family of distributions. If 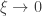 , it converges to Gumbel (type I), whereas if
, it converges to Gumbel (type I), whereas if 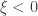 it corresponds to Fréchet (type II), and if
it corresponds to Fréchet (type II), and if  it corresponds to Weibull (type III). Inference on
it corresponds to Weibull (type III). Inference on 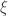 allows choice of a particular family for a given problem.
allows choice of a particular family for a given problem.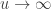 , the limiting distribution of a random variable
, the limiting distribution of a random variable  , conditional on
, conditional on 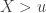 , is:
, is: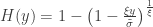
 and
and 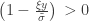 . The two parameters are the
. The two parameters are the 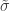 (scale). The shape corresponds to the same parameter
(scale). The shape corresponds to the same parameter 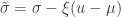 .
.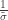 as
as ![\left[0, \tilde{\sigma}\right]](https://s0.wp.com/latex.php?latex=%5Cleft%5B0%2C+%5Ctilde%7B%5Csigma%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002) when
when 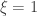 , and a Pareto distribution when
, and a Pareto distribution when  .
. , which represent distributions approximately exponential, parametters for the GPD can be estimated using at least three methods: maximum likelihood, moments, and probability-weighted moments. These are described in Hosking and Wallis (1987). For
, which represent distributions approximately exponential, parametters for the GPD can be estimated using at least three methods: maximum likelihood, moments, and probability-weighted moments. These are described in Hosking and Wallis (1987). For 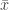 and
and 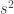 be respectively the sample mean and variance. The moment estimators of
be respectively the sample mean and variance. The moment estimators of  and
and  .
.

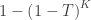
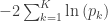

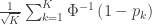
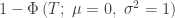
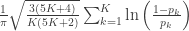

 is the number of partial tests, and the remaining of the variables follow the usual notation (see the Table 1 in the paper for the complete description). Many of these combining functions were proposed over the years for applications such as meta-analyses, and many of them assume independence between the tests being combined, and will give incorrect p-values if such assumption is not met. In the NPC, lack of dependence is not a problem, even if these same functions are used: the synchronised permutations ensure that any dependence, if existing, is taken into account, and this is done so implicitly, with no need for explicit modelling.
is the number of partial tests, and the remaining of the variables follow the usual notation (see the Table 1 in the paper for the complete description). Many of these combining functions were proposed over the years for applications such as meta-analyses, and many of them assume independence between the tests being combined, and will give incorrect p-values if such assumption is not met. In the NPC, lack of dependence is not a problem, even if these same functions are used: the synchronised permutations ensure that any dependence, if existing, is taken into account, and this is done so implicitly, with no need for explicit modelling.