
Pedigree-based analyses allow investigation of genetic and environmental influences on anatomy, physiology, and behaviour.
Methods based on components of variance have been used extensively to assess genetic influences and identify loci associated with various traits quantifying aspects of anatomy, physiology, and behaviour, in both normal and pathological conditions. In an earlier post, indices of genetic resemblance between relatives were presented, and in the last post, the kinship matrix was defined. In this post, these topics are used to present a basic model that allows partitioning of the phenotypic variance into sources of variation that can be ascribed to genetic, environmental, and other factors.
A simple model
Consider the model:

where, for  subjects,
subjects, 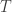 traits,
traits, 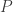 covariates and
covariates and 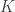 variance components,
variance components, 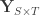 are the observed trait values for each subject,
are the observed trait values for each subject,  is a matrix of covariates,
is a matrix of covariates, 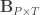 is a matrix of unknown covariates’ weights, and
is a matrix of unknown covariates’ weights, and  are the residuals after the covariates have been taken into account.
are the residuals after the covariates have been taken into account.
The elements of each column 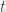 of
of  are assumed to follow a multivariate normal distribution
are assumed to follow a multivariate normal distribution  , where
, where  is the between-subject covariance matrix. The elements of each row
is the between-subject covariance matrix. The elements of each row  of are assumed to follow a normal distribution
of are assumed to follow a normal distribution 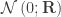 , where
, where  is the between-trait covariance matrix. Both and are seen as the sum of variance components, i.e.
is the between-trait covariance matrix. Both and are seen as the sum of variance components, i.e. 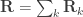 and
and 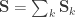 . For a discussion on these equalities, see Eisenhart (1947) [see references at the end].
. For a discussion on these equalities, see Eisenhart (1947) [see references at the end].
An equivalent model
The same model can be written in an alternative way. Let  be the stacked vector of traits,
be the stacked vector of traits,  is the matrix of covariates,
is the matrix of covariates,  the vector with the covariates’ weights,
the vector with the covariates’ weights,  the residuals after the covariates have been taken into account, and
the residuals after the covariates have been taken into account, and 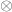 represent the Kronecker product. The model can then be written as:
represent the Kronecker product. The model can then be written as:

The stacked residuals  is assumed to follow a multivariate normal distribution
is assumed to follow a multivariate normal distribution  , where
, where  can be seen as the sum of variance components:
can be seen as the sum of variance components:

The here is the same as in Almasy and Blangero (1998). 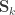 can be modelled as correlation matrices. The associated scalars are absorbed into the (to be estimated)
can be modelled as correlation matrices. The associated scalars are absorbed into the (to be estimated)  . is the phenotypic covariance matrix between the residuals of the traits:
. is the phenotypic covariance matrix between the residuals of the traits:
![\mathbf{R} = \left[ \begin{array}{ccc} \mathsf{Var}(\upsilon_1) & \cdots & \mathsf{Cov}(\upsilon_1,\upsilon_T) \\ \vdots & \ddots & \vdots \\ \mathsf{Cov}(\upsilon_T,\upsilon_1) & \cdots & \mathsf{Var}(\upsilon_T) \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7BR%7D+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BCov%7D%28%5Cupsilon_1%2C%5Cupsilon_T%29+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cmathsf%7BCov%7D%28%5Cupsilon_T%2C%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
whereas each are the share of these covariances attributable to the 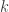 -th component:
-th component:
![\mathbf{R}_k = \left[ \begin{array}{ccccc} \mathsf{Var}_k(\upsilon_1) & \cdots & \mathsf{Cov}_k(\upsilon_1,\upsilon_T) \\ \vdots & \ddots & \vdots \\ \mathsf{Cov}_k(\upsilon_T,\upsilon_1) & \cdots & \mathsf{Var}_k(\upsilon_T) \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7BR%7D_k+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccccc%7D+%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BCov%7D_k%28%5Cupsilon_1%2C%5Cupsilon_T%29+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cmathsf%7BCov%7D_k%28%5Cupsilon_T%2C%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
can be converted to a between-trait phenotypic correlation matrix  as:
as:
![\mathbf{\mathring{R}} = \left[ \begin{array}{ccc} \frac{\mathsf{Var}(\upsilon_1)}{\mathsf{Var}(\upsilon_1)} & \cdots & \frac{\mathsf{Cov}(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}(\upsilon_1)\mathsf{Var}(\upsilon_T)\right)^{1/2}} \\ \vdots & \ddots & \vdots \\ \frac{\mathsf{Cov}(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}(\upsilon_1)\mathsf{Var}(\upsilon_T)\right)^{1/2}} & \cdots & \frac{\mathsf{Var}(\upsilon_T)}{\mathsf{Var}(\upsilon_T)} \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5Cmathring%7BR%7D%7D+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cfrac%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BCov%7D%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cfrac%7B%5Cmathsf%7BCov%7D%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%7D+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
The above phenotypic correlation matrix has unit diagonal and can still be fractioned into their components:
![\mathbf{\mathring{R}}_k = \left[ \begin{array}{ccc} \frac{\mathsf{Var}_k(\upsilon_1)}{\mathsf{Var}(\upsilon_1)} & \cdots & \frac{\mathsf{Cov}_k(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}(\upsilon_1)\mathsf{Var}(\upsilon_T)\right)^{1/2}} \\ \vdots & \ddots & \vdots \\ \frac{\mathsf{Cov}_k(\upsilon_T,\upsilon_1)}{\left(\mathsf{Var}(\upsilon_T)\mathsf{Var}(\upsilon_1)\right)^{1/2}} & \cdots & \frac{\mathsf{Var}_k(\upsilon_T)}{\mathsf{Var}(\upsilon_T)} \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5Cmathring%7BR%7D%7D_k+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_T%2C%5Cupsilon_1%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cright%29%5E%7B1%2F2%7D%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%7D+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
The relationship 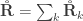 holds. The diagonal elements of
holds. The diagonal elements of  may receive particular names, e.g., heritability, environmentability, dominance effects, shared enviromental effects, etc., depending on what is modelled in the corresponding . However, the off-diagonal elements of are not the
may receive particular names, e.g., heritability, environmentability, dominance effects, shared enviromental effects, etc., depending on what is modelled in the corresponding . However, the off-diagonal elements of are not the 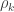 that correspond, e.g. to the genetic or environmental correlation. These off-diagonal elements are instead the signed
that correspond, e.g. to the genetic or environmental correlation. These off-diagonal elements are instead the signed  when
when 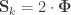 , or their
, or their  -equivalent for other variance components (see below). In this particular case, they can also be called “bivariate heritabilities” (Falconer and MacKay, 1996). A matrix
-equivalent for other variance components (see below). In this particular case, they can also be called “bivariate heritabilities” (Falconer and MacKay, 1996). A matrix  that contains these correlations , which are the fraction of the variance attributable to the -th component that is shared between pairs of traits is given by:
that contains these correlations , which are the fraction of the variance attributable to the -th component that is shared between pairs of traits is given by:
![\mathbf{\breve{R}}_k = \left[ \begin{array}{ccc} \frac{\mathsf{Var}_k(\upsilon_1)}{\mathsf{Var}_k(\upsilon_1)} & \cdots & \frac{\mathsf{Cov}_k(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}_k(\upsilon_1)\mathsf{Var}_k(\upsilon_T)\right)^{1/2}} \\ \vdots & \ddots & \vdots \\ \frac{\mathsf{Cov}_k(\upsilon_T,\upsilon_1)}{\left(\mathsf{Var}_k(\upsilon_T)\mathsf{Var}_k(\upsilon_1)\right)^{1/2}} & \cdots & \frac{\mathsf{Var}_k(\upsilon_T)}{\mathsf{Var}_k(\upsilon_T)} \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5Cbreve%7BR%7D%7D_k+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%7D%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_T%2C%5Cupsilon_1%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%5Cright%29%5E%7B1%2F2%7D%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%7D%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%7D+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
As for the phenotypic correlation matrix, each has unit diagonal.
The most common case
A particular case is when  , the coefficient of familial relationship between subjects, and
, the coefficient of familial relationship between subjects, and 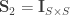 . In this case, the diagonal elements of
. In this case, the diagonal elements of  represent the heritability (
represent the heritability ( ) for each trait . The diagonal of
) for each trait . The diagonal of  contains
contains  , the environmentability. The off-diagonal elements of contain the signed (see below). The genetic correlations,
, the environmentability. The off-diagonal elements of contain the signed (see below). The genetic correlations,  are the off-diagonal elements of
are the off-diagonal elements of 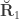 , whereas the off-diagonal elements of
, whereas the off-diagonal elements of 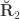 are
are 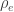 , the environmental correlations between traits. In this particular case, the components of , i.e., are equivalent to
, the environmental correlations between traits. In this particular case, the components of , i.e., are equivalent to 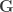 and
and 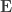 covariance matrices as in Almasy et al (1997).
covariance matrices as in Almasy et al (1997).
Relationship with the ERV
To see how the off-diagonal elements of are the signed Endophenotypic Ranking Values for each of the -th variance component, (Glahn et al, 2011), note that for a pair of traits 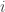 and
and 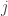 :
:

Multiplying both numerator and denominator by  and rearranging the terms gives:
and rearranging the terms gives:
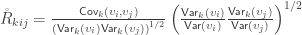
When 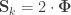 , the above reduces to
, the above reduces to  , which is the signed version of
, which is the signed version of  when is the genetic component.
when is the genetic component.
Positive-definiteness
and are covariance matrices and so, are positive-definite, whereas the correlation matrices , and are positive-semidefinite. A hybrid matrix that does not have to be positive-definite or semidefinite is:
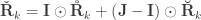
where 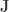 is a matrix of ones,
is a matrix of ones,  is the identity, both of size
is the identity, both of size 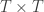 , and
, and 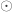 is the Hadamard product. An example of such matrix of practical use is to show concisely the heritabilities for each trait in the diagonal and the genetic correlations in the off-diagonal.
is the Hadamard product. An example of such matrix of practical use is to show concisely the heritabilities for each trait in the diagonal and the genetic correlations in the off-diagonal.
Cauchy-Schwarz
Algorithmic advantages can be obtained from the positive-definiteness of . The Cauchy–Schwarz theorem (Cauchy, 1821; Schwarz, 1888) states that:

Hence, the bounds for the off-diagonal elements can be known from the diagonal elements, which, by their turn, are estimated in a simpler, univariate model.

The Cauchy-Schwarz inequality imposes limits on the off-diagonal values of the matrix that contains the genetic covariances (or bivariate heritabilities).
Parameter estimation
Under the multivariate normal assumption, the parameters can be estimated maximising the following loglikelihood function:

where  is the number of observations on the stacked vector
is the number of observations on the stacked vector 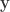 . Unbiased estimates for
. Unbiased estimates for  , although inefficient and inappropriate for hypothesis testing, can be obtained with ordinary least squares (OLS).
, although inefficient and inappropriate for hypothesis testing, can be obtained with ordinary least squares (OLS).
Parametric inference
Hypothesis testing can be performed with the likelihood ratio test (LRT), i.e., the test statistic is produced by subtracting from the loglikelihood of the model in which all the parameters are free to vary (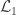 ), the loglikelihood of a model in which the parameters being tested are constrained to zero, the null model (
), the loglikelihood of a model in which the parameters being tested are constrained to zero, the null model (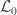 ). The statistic is given by
). The statistic is given by  (Wilks, 1938), which here is asymptotically distributed as a 50:50 mixture of a
(Wilks, 1938), which here is asymptotically distributed as a 50:50 mixture of a  and
and 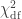 distributions, where df is the number of parameters being tested and free to vary in the unconstrained model (Self and Liang, 1987). From this distribution the p-values can be obtained.
distributions, where df is the number of parameters being tested and free to vary in the unconstrained model (Self and Liang, 1987). From this distribution the p-values can be obtained.
References
- Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998 May;62(5):1198–211.
- Almasy L, Dyer TD, Blangero J. Bivariate quantitative trait linkage analysis: pleiotropy versus co-incident linkages. Genet Epidemiol. 1997 Jan;14(6):953–8.
- Cauchy, A-L. Œuvres Completes D’Augustin Cauchy (published in 1882). Vol. XV. Gauthier-Villars et Fils, Paris, 1821.
- Eisenhart C. The assumptions underlying the analysis of variance. Biometrics. 1947;3(1):1–21.
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. Addison Wesley Longman, Harlow, Essex, UK, 1996.
- Glahn DC, Curran JE, Winkler AM, Carless MA, Kent JW, Charlesworth JC, et al. High dimensional endophenotype ranking in the search for major depression risk genes. Biol Psychiatry. 2012 Jan 1;71(1):6–14.
- Schwarz HA. Über ein Flächen kleinsten Flächeninhalts betreffendes Problem der Variationsrechnung. Acta Societatis Scientiarum Fennicæ XV, 319–332, 1888.
- Self SG, Liang KY. Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J Am Stat Assoc. 1987;82(398):605–10.
- Wilks SS. The Large-Sample Distribution of the Likelihood Ratio for Testing Composite Hypotheses. Ann Math Stat. 1938;9(1):60–2.
The photograph at the top (elephants) is by Anja Osenberg and was generously released into public domain.



Pingback: Understanding the kinship matrix | Brainder.