

Permutation tests are known to be superior to parametric tests: they are based on only few assumptions, essentially that the data are exchangeable, and allow the correction for the multiplicity of tests and the use of various non-standard statistics. The exchangeability assumption allows data to be permuted whenever their joint distribution remains unaltered. Usually this means that each observation needs to be independent from the others.
In many studies, however, there are repeated measurements on the same subjects, which violates exchangeability: clearly, the various measurements obtained from a given subject are not independent from each other. In the Human Connectome Project (HCP) (Van Essen et al, 2012; 2013; see references at the end), subjects are sampled along with their siblings (most of them are twins), such that independence cannot be guaranteed either.
In Winkler et al. (2014), certain structured types of non-independence in brain imaging were addressed through the definition of exchangeability blocks (EBs). Observations within EB can be shuffled freely or, alternatively, the EBs themselves can be shuffled as a whole. This allows various designs that otherwise could not be assessed through permutations.
The same idea can be generalised for blocks that are nested within other blocks, in a multi-level fashion. In the paper Multi-level Block Permutation (Winkler et al., 2015) we presented a method that allows blocks to be shuffled a whole, and inside them, sub-blocks are further allowed to be shuffled, in a recursive process. The method is flexible enough to accommodate permutations, sign-flippings (sometimes also called “wild bootstrap”), and permutations together with sign-flippings.
In particular, this permutation scheme allows the data of the HCP to be analysed via permutations: subjects are allowed to be shuffled with their siblings while keeping the joint distribution intra-sibship maintained. Then each sibship is allowed to be shuffled with others of the same type.
In the paper we show that the error type I is controlled at the nominal level, and the power is just marginally smaller than that would be obtained by permuting freely if free permutation were allowed. The more complex the block structure, the larger the reductions in power, although with large sample sizes, the difference is barely noticeable.
Importantly, simply ignoring family structure in designs as this causes the error rates not to be controlled, with excess false positives, and invalid results. We show in the paper examples of false positives that can arise, even after correction for multiple testing, when testing associations between cortical thickness, cortical area, and measures of body size as height, weight, and body-mass index, all of them highly heritable. Such false positives can be avoided with permutation tests that respect the family structure.
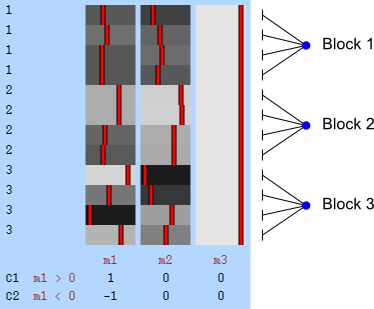
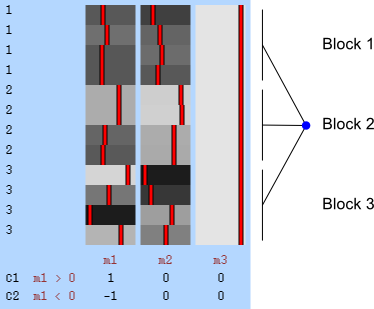
The figure at the top shows how the subjects of the HCP (terminal dots, shown in white colour) can be shuffled or not, while respecting the family structure. Blue dots indicate branches that can be permuted, whereas red dots indicate branches that cannot (see the main paper for details). This diagram includes 232 subjects of an early public release of HCP data. The tree on the left considers dizygotic twins as a category on their own, i.e., that cannot be shuffled with ordinary siblings, whereas the tree on the right considers dizygotic twins as ordinary siblings.
The first applied study using our strategy has just appeared. The method is implemented in the freely available package PALM — Permutation Analysis of Linear Models, and a set of practical steps to use it with actual HCP data is available here.
References
- Van Essen DC, Ugurbil K, Auerbach E, Barch D, Behrens TE, Bucholz R, Chang A, Chen L, Corbetta M, Curtiss SW, Della Penna S, Feinberg D, Glasser MF, Harel N, Heath AC, Larson-Prior L, Marcus D, Michalareas G, Moeller S, Oostenveld R, Petersen SE, Prior F, Schlaggar BL, Smith SM, Snyder AZ, Xu J, Yacoub E; WU-Minn HCP Consortium. The Human Connectome Project: a data acquisition perspective. Neuroimage. 2012;62(4):2222-31.
- Van Essen DC, Smith SM, Barch DM, Behrens TEJ, Yacoub E, Ugurbil K. The WU-Minn Human Connectome Project: An overview. Neuroimage. 2013;80:62-79.
- Winkler AM, Ridgway GR, Webster MA, Smith SM, Nichols TE. Permutation inference for the general linear model. Neuroimage. 2014;92:381-97. (Open Access)
- Winkler AM, Webster MA, Vidaurre D, Nichols TE, Smith SM. Multi-level block permutation. Neuroimage. 2015;123:253-68. (Open Access)
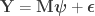
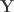 is the
is the 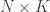 full rank matrix of observed data, with
full rank matrix of observed data, with 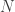 observations of
observations of 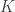 distinct (possibly non-independent) variables,
distinct (possibly non-independent) variables, 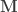 is the full-rank
is the full-rank  design matrix that includes explanatory variables (i.e., effects of interest and possibly nuisance effects),
design matrix that includes explanatory variables (i.e., effects of interest and possibly nuisance effects), 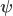 is the
is the  vector of
vector of 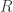 regression coefficients, and
regression coefficients, and  is the
is the 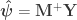 , where the superscript (
, where the superscript ( ) denotes a pseudo-inverse.
) denotes a pseudo-inverse. , where
, where 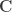 is a
is a  full-rank matrix of
full-rank matrix of  contrasts of coefficients on the regressors encoded in
contrasts of coefficients on the regressors encoded in  ,
,  and
and  is a
is a  full-rank matrix of
full-rank matrix of 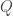 contrasts of coefficients on the dependent, response variables in
contrasts of coefficients on the dependent, response variables in  ; if
; if  or
or  , the model is univariate. Once the hypothesis has been established,
, the model is univariate. Once the hypothesis has been established, 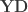 , such that the contrast
, such that the contrast 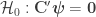 .
.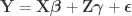
 is the matrix with nuisance regressors, and
is the matrix with nuisance regressors, and  and
and 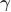 are respectively the vectors of regression coefficients. From this model we can also define the projection (hat) matrices
are respectively the vectors of regression coefficients. From this model we can also define the projection (hat) matrices  and
and 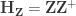 due to tue regressors of interest and nuisance, respectively, and the residual-forming matrices
due to tue regressors of interest and nuisance, respectively, and the residual-forming matrices  and
and  .
.![\left[ \mathbf{X} \; \mathbf{Z} \right]](https://s0.wp.com/latex.php?latex=%5Cleft%5B+%5Cmathbf%7BX%7D+%5C%3B+%5Cmathbf%7BZ%7D+%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002) , with
, with ![\boldsymbol{\psi} = \left[ \boldsymbol{\beta}' \; \boldsymbol{\gamma}' \right]'](https://s0.wp.com/latex.php?latex=%5Cboldsymbol%7B%5Cpsi%7D+%3D+%5Cleft%5B+%5Cboldsymbol%7B%5Cbeta%7D%27+%5C%3B+%5Cboldsymbol%7B%5Cgamma%7D%27+%5Cright%5D%27&bg=ffffff&fg=333333&s=0&c=20201002) . More involved strategies can, however, be devised to obtain some practical benefits. One such partitioning is to define
. More involved strategies can, however, be devised to obtain some practical benefits. One such partitioning is to define 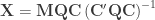 and
and , where
, where  ,
,  , and
, and 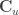 has
has  columns that span the null space of
columns that span the null space of ![[\mathbf{C} \; \mathbf{C}_u]](https://s0.wp.com/latex.php?latex=%5B%5Cmathbf%7BC%7D+%5C%3B+%5Cmathbf%7BC%7D_u%5D&bg=ffffff&fg=333333&s=0&c=20201002) is a
is a 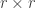 invertible, full-rank matrix (Smith et al, 2007). This partitioning has a number of features:
invertible, full-rank matrix (Smith et al, 2007). This partitioning has a number of features: 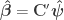 ,
,  , i.e., estimates and variances of
, i.e., estimates and variances of 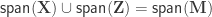 , i.e., the partitioned model spans the same space as the original.
, i.e., the partitioned model spans the same space as the original. and
and  . As with the previous strategy, the parameters of interest in the partitioned model are equal to the contrast of the original parameters. A full column rank nuisance partition can be obtained from the singular value decomposition (SVD) of
. As with the previous strategy, the parameters of interest in the partitioned model are equal to the contrast of the original parameters. A full column rank nuisance partition can be obtained from the singular value decomposition (SVD) of  .
.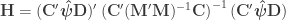
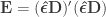
 and
and 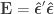 . The various well-known multivariate statistics (see
. The various well-known multivariate statistics (see 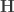 and
and 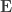 . Pillai’s trace is:
. Pillai’s trace is:
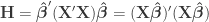
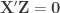 , and defining
, and defining  , we have:
, we have: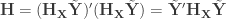

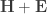 in the Pillai’s trace can therefore be rewritten as:
in the Pillai’s trace can therefore be rewritten as:

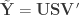 and
and  , where
, where  contains only the columns that correspond to non-zero eigenvalues. Thus, the above can be rewritten as:
contains only the columns that correspond to non-zero eigenvalues. Thus, the above can be rewritten as:
 .
. and
and  (or
(or  ), then
), then  . The first three moments of the permutation distribution of statistics that can be written in this form can be computed analytically once
. The first three moments of the permutation distribution of statistics that can be written in this form can be computed analytically once  and
and  are known. With the first three moments, a gamma distribution (Pearson type III) can be fit, thus allowing p-values to be computed without resorting to the usual beta approximation to Pillai’s trace, nor using permutations, yet with results that are not based on the assumption of normality (Mardia, 1971; Kazi-Aoual, 1995; Minas and Montana, 2014).
are known. With the first three moments, a gamma distribution (Pearson type III) can be fit, thus allowing p-values to be computed without resorting to the usual beta approximation to Pillai’s trace, nor using permutations, yet with results that are not based on the assumption of normality (Mardia, 1971; Kazi-Aoual, 1995; Minas and Montana, 2014).
 distinct possible orderings of these cups, and by telling the subject in advance that there are four cups of each type, this guarantees that the answer will include four of each.
distinct possible orderings of these cups, and by telling the subject in advance that there are four cups of each type, this guarantees that the answer will include four of each.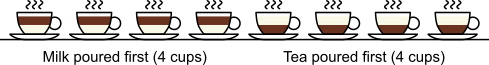
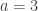
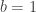


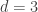
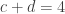
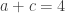
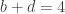
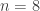
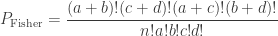
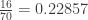 . This is not the final result, though: what matters to disprove the hypothesis that she is not able to discriminate is how likely it would be for her to find a result at least as extreme as the one observed. In this case, there is one case that is more extreme, which would be the one in which she would have made correct guesses for all the 8 cups, in which case the values in the contingency table above would have been
. This is not the final result, though: what matters to disprove the hypothesis that she is not able to discriminate is how likely it would be for her to find a result at least as extreme as the one observed. In this case, there is one case that is more extreme, which would be the one in which she would have made correct guesses for all the 8 cups, in which case the values in the contingency table above would have been 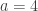 ,
,  ,
,  , and
, and  , with a probability computed with the same formula as
, with a probability computed with the same formula as  . Adding these two probabilities together yield
. Adding these two probabilities together yield  .
.
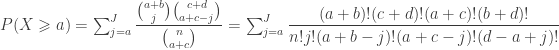
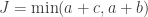 . Computing from the above (details omitted), yield the same value as using Fisher’s presentation, that is, the p-value is (exactly) 0.24286.
. Computing from the above (details omitted), yield the same value as using Fisher’s presentation, that is, the p-value is (exactly) 0.24286.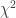 method
method
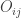 is the observed value for the element in the position
is the observed value for the element in the position 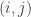 in the table,
in the table, 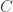 are respectively the number of rows and columns, and
are respectively the number of rows and columns, and 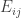 is the expected value for these elements if the null hypothesis is true. The values
is the expected value for these elements if the null hypothesis is true. The values  and column
and column  , divided by the overall number of observations
, divided by the overall number of observations  . A simpler, equivalent formula is given by:
. A simpler, equivalent formula is given by: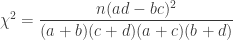
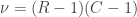 .
. , which corresponds to a p-value of 0.07865. However, it is well known that this method is inaccurate if cells in the table have too small quantities, usually below 5 or 6.
, which corresponds to a p-value of 0.07865. However, it is well known that this method is inaccurate if cells in the table have too small quantities, usually below 5 or 6.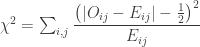
 , and a p-value of 0.23975, which is very similar to the one given by the Fisher method. Note again that this approach, like the
, and a p-value of 0.23975, which is very similar to the one given by the Fisher method. Note again that this approach, like the 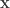 be a column vector containing binary indicators for whether milk was truly poured first. Let
be a column vector containing binary indicators for whether milk was truly poured first. Let 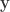 be a column vector containing binary indicators for whether the lady answered that milk was poured first. The
be a column vector containing binary indicators for whether the lady answered that milk was poured first. The  , where
, where  is a regression coefficient, and
is a regression coefficient, and 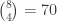 possible unique rearrangements. Out of these, in 17, there are 6 or more (out of 8) correct answers matching the values in
possible unique rearrangements. Out of these, in 17, there are 6 or more (out of 8) correct answers matching the values in 

 traits,
traits,  covariates and
covariates and 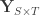 are the observed trait values for each subject,
are the observed trait values for each subject,  is a matrix of covariates,
is a matrix of covariates, 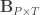 is a matrix of unknown covariates’ weights, and
is a matrix of unknown covariates’ weights, and  are the residuals after the covariates have been taken into account.
are the residuals after the covariates have been taken into account. of
of  are assumed to follow a multivariate normal distribution
are assumed to follow a multivariate normal distribution  , where
, where  is the between-subject covariance matrix. The elements of each row
is the between-subject covariance matrix. The elements of each row  of
of 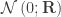 , where
, where  is the between-trait covariance matrix. Both
is the between-trait covariance matrix. Both 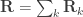 and
and 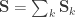 . For a discussion on these equalities, see Eisenhart (1947) [see references at the end].
. For a discussion on these equalities, see Eisenhart (1947) [see references at the end]. be the stacked vector of traits,
be the stacked vector of traits,  is the matrix of covariates,
is the matrix of covariates,  the vector with the covariates’ weights,
the vector with the covariates’ weights,  the residuals after the covariates have been taken into account, and
the residuals after the covariates have been taken into account, and 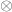 represent the
represent the 
 is assumed to follow a multivariate normal distribution
is assumed to follow a multivariate normal distribution  , where
, where  can be seen as the sum of
can be seen as the sum of 
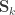 can be modelled as correlation matrices. The associated scalars are absorbed into the (to be estimated)
can be modelled as correlation matrices. The associated scalars are absorbed into the (to be estimated)  .
. ![\mathbf{R} = \left[ \begin{array}{ccc} \mathsf{Var}(\upsilon_1) & \cdots & \mathsf{Cov}(\upsilon_1,\upsilon_T) \\ \vdots & \ddots & \vdots \\ \mathsf{Cov}(\upsilon_T,\upsilon_1) & \cdots & \mathsf{Var}(\upsilon_T) \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7BR%7D+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BCov%7D%28%5Cupsilon_1%2C%5Cupsilon_T%29+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cmathsf%7BCov%7D%28%5Cupsilon_T%2C%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
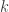 -th component:
-th component:![\mathbf{R}_k = \left[ \begin{array}{ccccc} \mathsf{Var}_k(\upsilon_1) & \cdots & \mathsf{Cov}_k(\upsilon_1,\upsilon_T) \\ \vdots & \ddots & \vdots \\ \mathsf{Cov}_k(\upsilon_T,\upsilon_1) & \cdots & \mathsf{Var}_k(\upsilon_T) \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7BR%7D_k+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccccc%7D+%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BCov%7D_k%28%5Cupsilon_1%2C%5Cupsilon_T%29+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cmathsf%7BCov%7D_k%28%5Cupsilon_T%2C%5Cupsilon_1%29+%26+%5Ccdots+%26+%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
 as:
as:![\mathbf{\mathring{R}} = \left[ \begin{array}{ccc} \frac{\mathsf{Var}(\upsilon_1)}{\mathsf{Var}(\upsilon_1)} & \cdots & \frac{\mathsf{Cov}(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}(\upsilon_1)\mathsf{Var}(\upsilon_T)\right)^{1/2}} \\ \vdots & \ddots & \vdots \\ \frac{\mathsf{Cov}(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}(\upsilon_1)\mathsf{Var}(\upsilon_T)\right)^{1/2}} & \cdots & \frac{\mathsf{Var}(\upsilon_T)}{\mathsf{Var}(\upsilon_T)} \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5Cmathring%7BR%7D%7D+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cfrac%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BCov%7D%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cfrac%7B%5Cmathsf%7BCov%7D%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%7D+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
![\mathbf{\mathring{R}}_k = \left[ \begin{array}{ccc} \frac{\mathsf{Var}_k(\upsilon_1)}{\mathsf{Var}(\upsilon_1)} & \cdots & \frac{\mathsf{Cov}_k(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}(\upsilon_1)\mathsf{Var}(\upsilon_T)\right)^{1/2}} \\ \vdots & \ddots & \vdots \\ \frac{\mathsf{Cov}_k(\upsilon_T,\upsilon_1)}{\left(\mathsf{Var}(\upsilon_T)\mathsf{Var}(\upsilon_1)\right)^{1/2}} & \cdots & \frac{\mathsf{Var}_k(\upsilon_T)}{\mathsf{Var}(\upsilon_T)} \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5Cmathring%7BR%7D%7D_k+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_T%2C%5Cupsilon_1%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%5Cmathsf%7BVar%7D%28%5Cupsilon_1%29%5Cright%29%5E%7B1%2F2%7D%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%7D%7B%5Cmathsf%7BVar%7D%28%5Cupsilon_T%29%7D+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
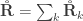 holds. The diagonal elements of
holds. The diagonal elements of  may receive particular names, e.g., heritability, environmentability, dominance effects, shared enviromental effects, etc., depending on what is modelled in the corresponding
may receive particular names, e.g., heritability, environmentability, dominance effects, shared enviromental effects, etc., depending on what is modelled in the corresponding 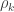 that correspond, e.g. to the genetic or environmental correlation. These off-diagonal elements are instead the signed
that correspond, e.g. to the genetic or environmental correlation. These off-diagonal elements are instead the signed  when
when 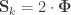 , or their
, or their  -equivalent for other variance components (see below). In this particular case, they can also be called “bivariate heritabilities” (Falconer and MacKay, 1996). A matrix
-equivalent for other variance components (see below). In this particular case, they can also be called “bivariate heritabilities” (Falconer and MacKay, 1996). A matrix  that contains these correlations
that contains these correlations ![\mathbf{\breve{R}}_k = \left[ \begin{array}{ccc} \frac{\mathsf{Var}_k(\upsilon_1)}{\mathsf{Var}_k(\upsilon_1)} & \cdots & \frac{\mathsf{Cov}_k(\upsilon_1,\upsilon_T)}{\left(\mathsf{Var}_k(\upsilon_1)\mathsf{Var}_k(\upsilon_T)\right)^{1/2}} \\ \vdots & \ddots & \vdots \\ \frac{\mathsf{Cov}_k(\upsilon_T,\upsilon_1)}{\left(\mathsf{Var}_k(\upsilon_T)\mathsf{Var}_k(\upsilon_1)\right)^{1/2}} & \cdots & \frac{\mathsf{Var}_k(\upsilon_T)}{\mathsf{Var}_k(\upsilon_T)} \end{array}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7B%5Cbreve%7BR%7D%7D_k+%3D+%5Cleft%5B+%5Cbegin%7Barray%7D%7Bccc%7D+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%7D%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_1%2C%5Cupsilon_T%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%5Cright%29%5E%7B1%2F2%7D%7D+%5C%5C+%5Cvdots+%26+%5Cddots+%26+%5Cvdots+%5C%5C+%5Cfrac%7B%5Cmathsf%7BCov%7D_k%28%5Cupsilon_T%2C%5Cupsilon_1%29%7D%7B%5Cleft%28%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%5Cmathsf%7BVar%7D_k%28%5Cupsilon_1%29%5Cright%29%5E%7B1%2F2%7D%7D+%26+%5Ccdots+%26+%5Cfrac%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%7D%7B%5Cmathsf%7BVar%7D_k%28%5Cupsilon_T%29%7D+%5Cend%7Barray%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002)
 , the coefficient of familial relationship between subjects, and
, the coefficient of familial relationship between subjects, and 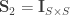 . In this case, the
. In this case, the  represent the heritability (
represent the heritability ( ) for each trait
) for each trait  contains
contains  , the environmentability. The off-diagonal elements of
, the environmentability. The off-diagonal elements of  are the off-diagonal elements of
are the off-diagonal elements of 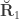 , whereas the off-diagonal elements of
, whereas the off-diagonal elements of 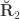 are
are 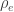 , the environmental correlations between traits. In this particular case, the components of
, the environmental correlations between traits. In this particular case, the components of 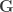 and
and 
 and rearranging the terms gives:
and rearranging the terms gives: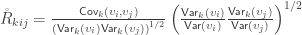
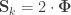 , the above reduces to
, the above reduces to  , which is the signed version of
, which is the signed version of  when
when 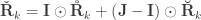
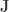 is a matrix of ones,
is a matrix of ones,  is the identity, both of size
is the identity, both of size 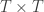 , and
, and 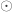 is the
is the 


 is the number of observations on the stacked vector
is the number of observations on the stacked vector 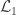 ), the loglikelihood of a model in which the parameters being tested are constrained to zero, the null model (
), the loglikelihood of a model in which the parameters being tested are constrained to zero, the null model (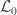 ). The statistic is given by
). The statistic is given by  (Wilks, 1938), which here is asymptotically distributed as a 50:50 mixture of a
(Wilks, 1938), which here is asymptotically distributed as a 50:50 mixture of a  and
and 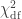 distributions, where df is the number of parameters being tested and free to vary in the unconstrained model (Self and Liang, 1987). From this distribution the p-values can be obtained.
distributions, where df is the number of parameters being tested and free to vary in the unconstrained model (Self and Liang, 1987). From this distribution the p-values can be obtained. , is probably the most interesting. It gives a probabilistic estimate that a random gene from a given subject
, is probably the most interesting. It gives a probabilistic estimate that a random gene from a given subject  matrix termed kinship matrix, usually represented as
matrix termed kinship matrix, usually represented as  , that has elements
, that has elements 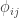 , and that can be used to model the covariance between individuals in quantitative genetics.
, and that can be used to model the covariance between individuals in quantitative genetics.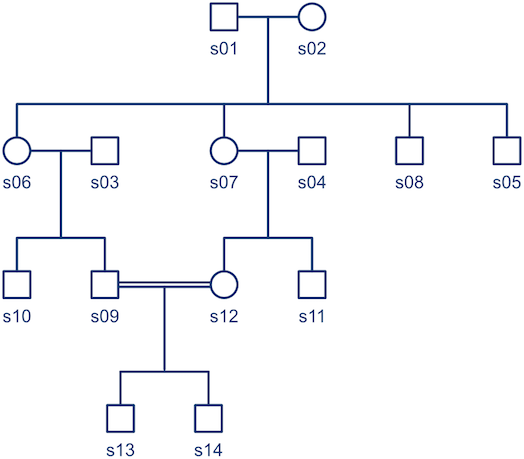
 ), is:
), is: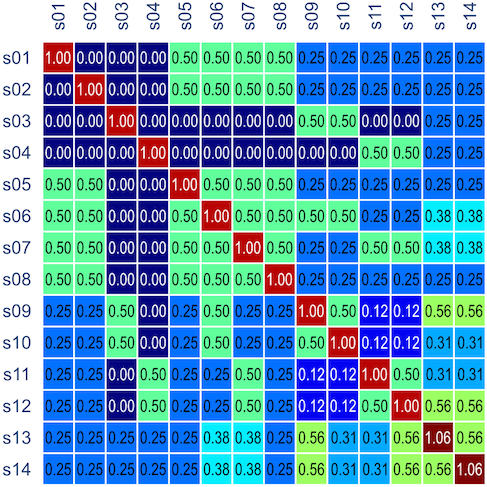

 . Each individual has two copies, one from paternal, another from maternal origin; these can be indicated as
. Each individual has two copies, one from paternal, another from maternal origin; these can be indicated as  and
and  for individual
for individual 
 , a respective probability
, a respective probability  can be assigned; these are called coefficients of identity by descent. These probabilities can be calculated at every generation following very elementary rules. For most problems, however, the distinction between paternal and maternal origin of a gene is irrelevant, and some of the above states are equivalent to others. If these are condensed, we can retain 9 distinct ways, shown in the figure below:
can be assigned; these are called coefficients of identity by descent. These probabilities can be calculated at every generation following very elementary rules. For most problems, however, the distinction between paternal and maternal origin of a gene is irrelevant, and some of the above states are equivalent to others. If these are condensed, we can retain 9 distinct ways, shown in the figure below:
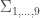 , a respective probability
, a respective probability 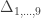 can be assigned; these are called condensed coefficients of identity by descent, and relate to the former as:
can be assigned; these are called condensed coefficients of identity by descent, and relate to the former as: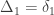
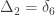

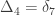




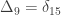
 ,
,  and
and 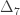 correspond to his coefficients
correspond to his coefficients  ,
,  and
and  .
.
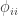 is the kinship of a subject with himself. Two genes taken from the same individual can either be the same gene (probability
is the kinship of a subject with himself. Two genes taken from the same individual can either be the same gene (probability  of being the same) or be the genes inherited from father and mother, in which case the probability is given by the coefficient of kinship between the parents. In other words,
of being the same) or be the genes inherited from father and mother, in which case the probability is given by the coefficient of kinship between the parents. In other words,  . If both parents are unrelated,
. If both parents are unrelated, 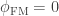 , such that the kinship of a subject with himself is
, such that the kinship of a subject with himself is 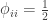 .
. (see below about the coefficient of inbreeding,
(see below about the coefficient of inbreeding, 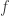 ). Thus, if there are
). Thus, if there are 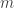 generations between
generations between  . If
. If 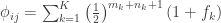
 , and used to model the covariance between subjects as
, and used to model the covariance between subjects as  (
(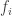 can be computed from the coefficients of identity:
can be computed from the coefficients of identity:

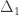

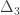
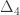

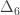
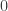
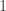

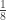
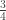

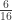






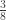
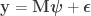
 vector of observations,
vector of observations,  matrix of explanatory variables,
matrix of explanatory variables,  vector of regression coefficients, and
vector of regression coefficients, and  , where
, where  matrix that defines a contrast of regression coefficients, satisfying
matrix that defines a contrast of regression coefficients, satisfying 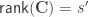 and
and  .
.
 vector of observations,
vector of observations, 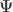 is the
is the 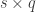 vector of regression coefficients, and
vector of regression coefficients, and  , where
, where  matrix that defines a contrast of observed variables, satisfying
matrix that defines a contrast of observed variables, satisfying  and
and 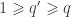 .
.
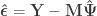
 and
and 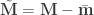 .
.
 , the multiple correlation coefficient can be computed from the
, the multiple correlation coefficient can be computed from the 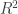 statistic as:
statistic as: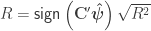
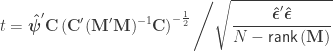
 statistic can be computed as:
statistic can be computed as: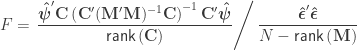
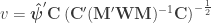
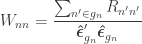
 are the
are the  diagonal elements of the residual forming matrix
diagonal elements of the residual forming matrix  , and
, and 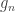 is the variance group to which the
is the variance group to which the 
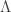 , is given by:
, is given by: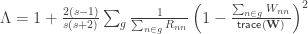

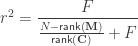
 is known, the formula can be solved for
is known, the formula can be solved for 

![\mathbf{A} = \left[\mathbf{y}\; \mathbf{M}\right]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7BA%7D+%3D+%5Cleft%5B%5Cmathbf%7By%7D%5C%3B+%5Cmathbf%7BM%7D%5Cright%5D&bg=ffffff&fg=333333&s=0&c=20201002) , and
, and  be the inverse of the correlation matrix of the columns of
be the inverse of the correlation matrix of the columns of 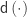 the diagonal operator, that returns a column vector with the diagonal entries of a square matrix. Then the matrix with the partial correlations is:
the diagonal operator, that returns a column vector with the diagonal entries of a square matrix. Then the matrix with the partial correlations is:
 ” is taken elementwise (i.e., not matrix power).
” is taken elementwise (i.e., not matrix power).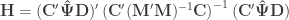 as the sums of products of the hypothesis. In fact, the original model can be modified as
as the sums of products of the hypothesis. In fact, the original model can be modified as  , where
, where 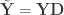 ,
, 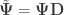 and
and  .
. , this is an univariate model, otherwise it remains multivariate, although
, this is an univariate model, otherwise it remains multivariate, although  .
.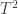 statistic can be computed as:
statistic can be computed as:


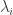 be the eigenvalues of
be the eigenvalues of 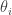 the eigenvalues of
the eigenvalues of 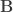 . Then:
. Then: .
. .
. .
.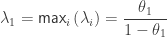 (analogous to
(analogous to  (analogous to
(analogous to  is the
is the  , where
, where 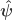 and
and 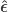 indicate that these are quantities estimated from the sample. If
indicate that these are quantities estimated from the sample. If 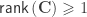 , the F statistic can be obtained as:
, the F statistic can be obtained as: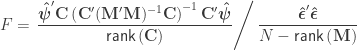
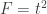 . For either of these statistics, we can assess their significance by repeating the same fit after permuting
. For either of these statistics, we can assess their significance by repeating the same fit after permuting 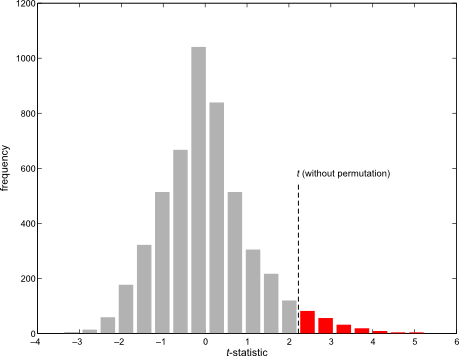
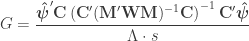
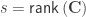 . The matrix
. The matrix 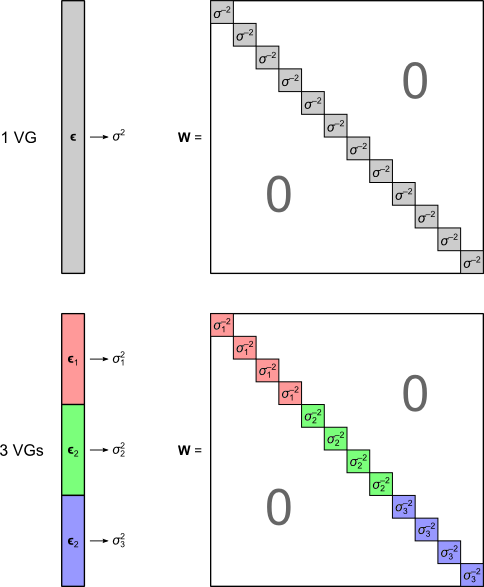
 , the t-equivalent to the G-statistic is
, the t-equivalent to the G-statistic is  -statistic for the Behrens-Fisher problem. The relationship between
-statistic for the Behrens-Fisher problem. The relationship between 

 and
and  .
.

 is the estimated p-value,
is the estimated p-value,  is the statistic for the unpermuted model,
is the statistic for the unpermuted model,  is the statistic for the
is the statistic for the  is an indicator function that evaluates as 1 if the condition between parenthesis is true, or 0 otherwise. This formulation produces unbiased results: since
is an indicator function that evaluates as 1 if the condition between parenthesis is true, or 0 otherwise. This formulation produces unbiased results: since  , the true p-value, then
, the true p-value, then  .
. , is small, or if the number of permutations is not sufficiently large, even after all the
, is small, or if the number of permutations is not sufficiently large, even after all the  may reach or surpass the value of
may reach or surpass the value of 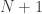 :
:
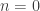 ,
,  , and so,
, and so, 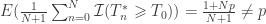 .
. , in a way that no p-value smaller than
, in a way that no p-value smaller than 
 be the observed values, then:
be the observed values, then:
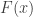 is the empirical cdf,
is the empirical cdf,  is the probability of observing a value in
is the probability of observing a value in 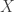 that is smaller than or equal to
that is smaller than or equal to  ,
,  is a function that evaluates as 1 if the condition in the parenthesis is satisfied or 0 otherwise, and
is a function that evaluates as 1 if the condition in the parenthesis is satisfied or 0 otherwise, and 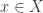 can be computed as
can be computed as  . For a discrete cdf, however, this trivial relationship does not hold. The reason is that, while for continuous distributions the probability
. For a discrete cdf, however, this trivial relationship does not hold. The reason is that, while for continuous distributions the probability  , for discrete distributions (such as empirical distributions),
, for discrete distributions (such as empirical distributions),  whenever
whenever 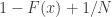 .
. . Then
. Then  , where
, where 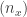 represents the ascending rank of
represents the ascending rank of 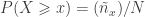 , where
, where  represents the descending rank of
represents the descending rank of 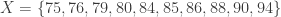

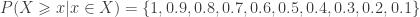

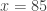 would be either 0.1, 0.2 or 0.3, depending on which instance of the “85” we were to choose. The correct value, however, is 0.3 (only), since 3 out of 10 variables are equal or above 85. The solution to this problem is to replace the simple, ordinal ranking, for a modified version of the so called competition ranking.
would be either 0.1, 0.2 or 0.3, depending on which instance of the “85” we were to choose. The correct value, however, is 0.3 (only), since 3 out of 10 variables are equal or above 85. The solution to this problem is to replace the simple, ordinal ranking, for a modified version of the so called competition ranking.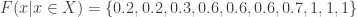
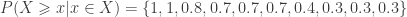
 , with
, with 